
¿Qué es la galactosemia y por qué ocurre en un niño? Síntomas, diagnóstico de la enfermedad. Tratamiento de la galactosemia en recién nacidos y medidas preventivas disponibles.
El contenido del artículo:- ¿Qué es la galactosemia?
- Síntomas del desarrollo
- Diagnóstico
- Cómo tratar la galactosemia
- Prevención
La galactosemia es una anomalía hereditaria rara pero aún común asociada con el metabolismo. La enzimopatía se asocia con una disfunción del metabolismo de los carbohidratos, como resultado de lo cual el cuerpo acumula galactosa y sus derivados. Cantidades excesivas de galactosa conducen al desarrollo de un cuadro clínico. En el caso de un diagnóstico tardío de galactosemia, los síntomas se vuelven pronunciados y el pronóstico de normalización de la condición del paciente es sombrío. Las parejas con predisposición a la enfermedad deben conocer los tipos de galactosemia, los métodos de diagnóstico, tratamiento y prevención, y también pensar en la planificación familiar. Las medidas tomadas a tiempo reducen significativamente el riesgo de complicaciones en el recién nacido.
¿Qué es la galactosemia?
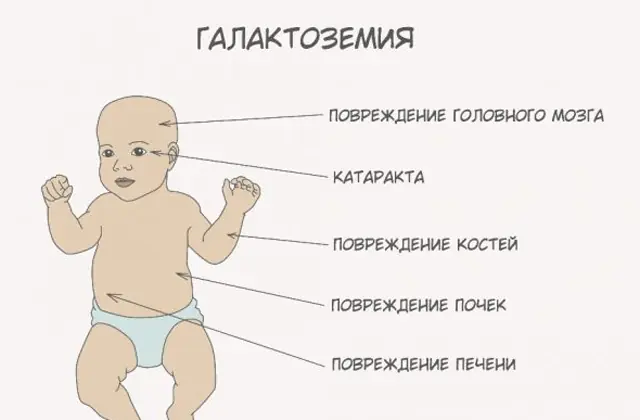
Antes de hablar de recomendaciones para la galactosemia, es necesario comprender el papel de la galactosa en el metabolismo de los carbohidratos. Este azúcar simple ingresa al cuerpo humano como parte de la lactosa, un disacárido cuyo segundo componente es la glucosa. Normalmente, el monosacárido se utiliza como recurso "energético" para células y compuestos complejos, materiales de "construcción" del tejido nervioso, membranas y terminaciones nerviosas.
En una persona sana, la lactosa se descompone en sus componentes en el tracto digestivo y se procesa gradualmente. Así, en la primera etapa de procesamiento, se obtiene galactosa 1-fosfato bajo la acción de una enzima especial galactoquinasa y ATP. En la siguiente etapa, como resultado del intercambio bajo la acción de enzimas, se obtienen glucosa 1-fosfato y uridina fosfato galactosa. Los monosacáridos se absorben mucho más fácilmente en los intestinos durante la digestión, lo cual es muy importante para los recién nacidos, ya que la leche es la base de su dieta.
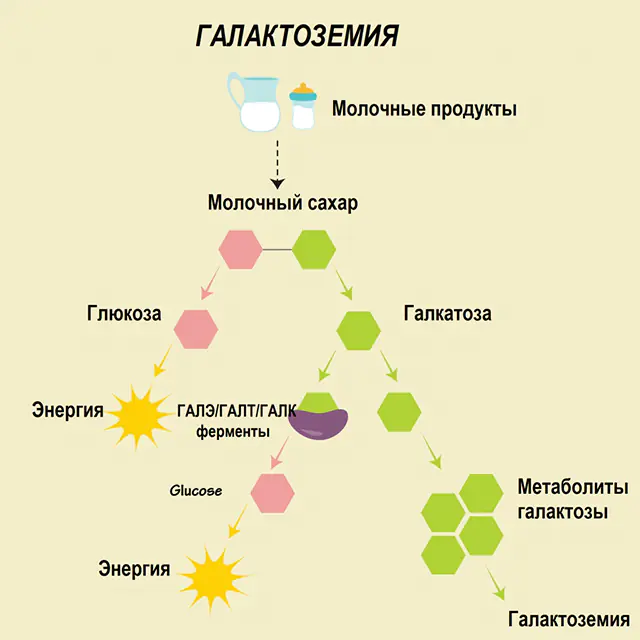
En los procesos de procesamiento participan tres enzimas: GALA (galactoquinasa), GALT (galactosa 1-fosfato uridiil transferasa), GALE (epimerasa). La producción deficiente de al menos una de esas enzimas en la primera o segunda etapa del metabolismo de los carbohidratos conduce a la acumulación de metabolitos, intoxicación del cuerpo y la posterior manifestación de síntomas de galactosemia.
Dependiendo de la enzima defectuosa, se distinguen los tipos correspondientes de galactosemia:
- Violación GALT- el primer tipo ocurre en un caso por cada 40 mil recién nacidos, el análisis de galactosemia mostrará una actividad enzimática reducida;
- patología GALC- el segundo tipo ocurre en un niño de cada 500 mil, mientras que la actividad de la enzima básica en los eritrocitos está dentro del rango normal, pero los síntomas de la enfermedad son pronunciados;
- VENDAVAL— el tercer tipo es el que menos casos se registra (un caso por millón de personas) y se caracteriza por síntomas leves.
El tipo más común de enfermedad (GALT) se divide en formas, según la genética de la galactosemia:
- La falla es característica de la etapa de transformación de la amida del ácido aspártico en aspartato; la patología también se llama signo de Duarte (Duarte). Es de destacar que una persona con tal anomalía se sentirá absolutamente sana, aunque la actividad de la enzima GALT se reduce entre un 25% y un 50%.
- La falla en la transición glutamina-arginina se registra con mayor frecuencia en caucásicos; la actividad enzimática es solo el 10% de lo normal, lo que provoca un curso grave de la enfermedad.
- Se produce una interrupción de la transición arginina-triptófano cuando la enzima está completamente inactiva, una forma extremadamente grave de la enfermedad.
- La interrupción del proceso de transformación lisina-asparagina ocurre con bastante frecuencia.
- La falla de la fermentación de serina-leucina se registró solo en representantes de la raza negroide y se asocia con una actividad enzimática insuficiente en el hígado (a un nivel de no más del 10% de la norma generalmente aceptada).
La enzima GALT está presente en el organismo en cantidades excesivas, por lo que una ligera disminución de su actividad no siempre se manifiesta como síntomas clínicos. Sólo cuando la actividad de GALT disminuya en un 50 por ciento o más aparecerán los primeros signos de la enfermedad.
La intoxicación del cuerpo con galactosa y metabolitos es peligrosa para el desarrollo de complicaciones a largo plazo: deterioro de la función motora, del habla, de las funciones reproductivas, especialmente en las niñas (a pesar de que la frecuencia de enfermedades en niños y niñas es la misma), Retraso del crecimiento y otras disfunciones del desarrollo.
¡Nota! La patogénesis de la enfermedad aún no se ha estudiado completamente. Sin embargo, ya se ha establecido que la acumulación de galactosa en el organismo es peligrosa no sólo por su dosis tóxica, sino también por su efecto inhibidor sobre otras enzimas. Sin el tratamiento y control adecuados, el paciente puede desarrollar un síndrome hipoglucémico.Como ya se señaló, la patología es una anomalía congénita. El tipo de herencia de la galactosemia es autosómica recesiva, es decir, el niño recibe genes defectuosos de los padres. El gen mutado se puede encontrar en diferentes alelos (formas de la afección): defectuoso y normal, cuando el defecto se transmitió de un solo padre. En este caso, la enfermedad se manifiesta en menor medida. Pero si los alelos defectuosos se transmiten tanto del padre como de la madre de acuerdo con el tipo de herencia de la galactosemia, los primeros signos de la enfermedad aparecerán unos días después del nacimiento del bebé. Es de destacar que si se diagnostica una enfermedad en un niño de una familia, la probabilidad de que la patología ocurra en el siguiente hijo de los mismos padres aumenta en un 25%.
La producción insuficiente de enzimas conduce a la acumulación de galactosa en el cuerpo y, como resultado, aparecen signos de galactosemia y se alteran las funciones del sistema excretor. Sin tratamiento, a los pocos meses de vida, una persona muere por insuficiencia hepática o infecciones que afectan el cuerpo.
Es importante que las principales causas de la galactosemia (componente hereditario) puedan establecerse incluso antes del nacimiento del niño, pero los médicos no excluyen la influencia de otros indicadores en el desarrollo de la patología:
- otras mutaciones genéticas;
- tejido afectado del hígado y sistema nervioso central;
- acumulación de líquido en el cristalino del ojo, visión borrosa.
Y, sin embargo, si a un paciente se le diagnostica galactosemia, la genética juega un papel clave. Los factores concomitantes son importantes sólo en pacientes heterocigotos, es decir, aquellos en quienes el gen defectuoso fue transmitido por uno de los padres. En pacientes homocigotos, la patología en sí es grave y requiere un tratamiento altamente especializado.
Síntomas del desarrollo de galactosemia.
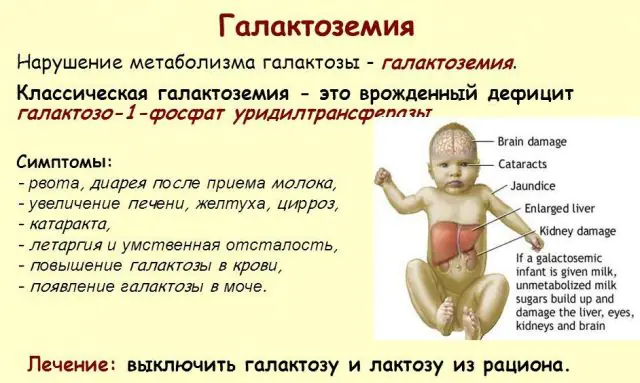
Según la gravedad de las manifestaciones de los signos de galactosemia, se distinguen tres grados de la enfermedad:
- luz- normalmente se detecta accidentalmente debido a una intolerancia a la leche;
- promedio- los primeros signos aparecen sólo después de beber leche;
- pesado— los síntomas de la patología se superponen a otras disfunciones del organismo (acumulación de líquido en la cavidad abdominal, sepsis).
La patología GALT clásica se manifiesta en su forma más grave. Los primeros signos de la enfermedad se harán visibles después del nacimiento del niño y de su primera toma, ya que la leche constituye la dieta principal del recién nacido. Después de alimentarse, experimenta vómitos, deposiciones frecuentes y aumento de la somnolencia en un contexto de letargo constante e hipotensión muscular. A pesar de la lactancia materna regular, no se produce ningún aumento de peso. Los trastornos digestivos son uno de los primeros signos de disfunción.
A medida que el cuerpo se intoxica, los signos de daño hepático se vuelven pronunciados: ictericia, agrandamiento del hígado. En unas pocas semanas, el niño desarrolla cataratas y, después de unos meses, debido a la intoxicación del tejido nervioso, se observan alteraciones en las funciones psicomotoras. La actividad de los túbulos renales se altera, como resultado de lo cual aparece azúcar reductor en la orina y se observa una disminución de la coagulación sanguínea.
Sin la terapia necesaria, los síntomas empeoran a medida que el cuerpo del bebé se intoxica y aumenta el riesgo para el desarrollo normal e incluso la vida humana. Del 20 al 30% de los niños, si se rechaza el tratamiento o si la enfermedad se diagnostica tarde, mueren a causa de la sepsis, que se desarrolla debido a la actividad leucocitosa. Los que sobreviven padecen insuficiencia renal y problemas de desarrollo psicomotor.
En muchos sentidos, el grado de manifestación de la patología depende de la causa hereditaria. La galactosemia se manifiesta cuando se produce una deficiencia de GALT. Así, el signo de Duhart al principio sólo puede manifestarse como ictericia prolongada (hasta 2 meses después del nacimiento), las cataratas y la disfunción hepática se diagnostican un poco más tarde. Pero si la actividad de la enzima GALT es al menos del 50%, es posible que no aparezcan síntomas clínicos.
La falta de enzima GALA no es tan pronunciada. El síntoma principal de la enfermedad son las cataratas progresivas y con el tiempo también pueden aparecer monosacáridos en la orina. Al mismo tiempo, la pérdida de peso del bebé es insignificante y sus habilidades psicomotoras se desarrollan dentro de los límites normales.
La deficiencia de GALE es inicialmente extremadamente rara, pero los síntomas aún se dividen en dos formas: benigna, cuando la falta de la enzima afecta a las células sanguíneas, y generalizada, cuando la deficiencia afecta a todos los tejidos del cuerpo. En el primer caso, sólo un análisis de galactosemia ayudará a detectar la enfermedad en las primeras etapas, sólo con el tiempo se notará una disfunción de las habilidades psicomotoras y del desarrollo del habla. La segunda forma es similar en sus síntomas a la deficiencia de GALT, pero en este caso también hay un agrandamiento del bazo. Incluso con el tratamiento de una forma grave de la enfermedad GALE, con el tiempo el niño experimenta retraso en el desarrollo psicomotor, sordera y problemas de visión.
Diagnóstico de galactosemia.
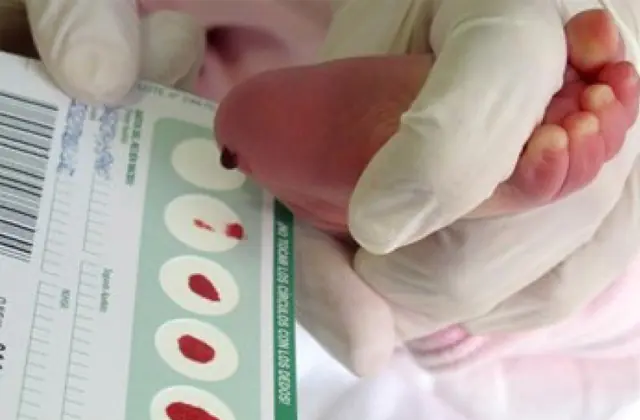
El cribado neonatal, introducido en varios países, permite identificar hasta 50 enfermedades congénitas en las primeras etapas, incluida la producción insuficiente de enzimas que participan activamente en el metabolismo. Si por algún motivo no se realiza el cribado, la enfermedad se identifica en función de características agregadas.
Por lo tanto, si se sospecha galactosemia, las recomendaciones del médico se hacen sólo después de un diagnóstico detallado. Un paciente pequeño debe someterse a:
- examen general: si hay signos de ictericia;
- se evalúan los signos generales: la cantidad y calidad de las deposiciones, la presencia o ausencia de hipotonicidad muscular, problemas con las habilidades psicomotoras;
- pruebas de laboratorio: análisis de sangre para bioquímica, análisis de sangre general, análisis de orina para detectar azúcar y proteínas;
- examen adicional del estado del hígado y los riñones.
Los diagnósticos de ADN son obligatorios para detectar mutaciones en GALT. Si el niño presenta síntomas de la enfermedad y la actividad de las enzimas GALT según los resultados de las pruebas es normal, se realiza un diagnóstico de ADN para la enzima GALE.
Los médicos pueden prescribir una serie de pruebas adicionales para diferenciar el diagnóstico. Es necesario un diagnóstico exclusivo para confirmar o refutar la presencia de diabetes mellitus y otras patologías asociadas a niveles elevados de azúcar, así como enfermedades que provocan agrandamiento del hígado.
¿Cómo tratar la galactosemia?

Una vez que el cribado neonatal y otros exámenes han confirmado la presencia de galactosemia, se prescribe el tratamiento. Es necesario comprender que, dado que la enfermedad es causada por una predisposición genética, una cura completa en esta etapa del desarrollo médico es imposible. La terapia tiene como objetivo reducir las manifestaciones clínicas de la patología, minimizando el progreso de la enfermedad y el desarrollo de complicaciones.
El principal método de tratamiento es una nutrición adecuada para la galactosemia. Todos los productos que contienen leche, incluso pan, dulces, embutidos y otros, quedan completamente excluidos de la dieta del paciente. También se eliminan los productos de origen vegetal y animal que contengan oligosacáridos: legumbres, soja, espinacas, chocolate, frutos secos, hígado, sesos, huevos y derivados de estos productos. La terapia dietética estricta es la única forma de prevenir la intoxicación del cuerpo.
Para los recién nacidos, la nutrición para la galactosemia también está especializada. La leche materna se elimina progresivamente por completo de la dieta y se sustituye por fórmulas sin lactosa y formulaciones a base de aislado de proteína de soja. La cantidad de ingredientes nutricionales en este caso debe corresponder a los estándares para un niño sano. Si un bebé presenta síntomas de alergia al aislado de proteína de soja, se seleccionan mezclas a base de hidrolizados de caseína. La primera ingesta de mezcla de alimentos debe ser 1/10 del requerimiento nutricional diario del niño. En 5 a 7 días, la leche materna se elimina por completo de la dieta.
Los primeros alimentos complementarios para niños con patología se prescriben a partir de los 4 meses. La dieta se amplía con zumos de frutas (manzana, pera y otros). Al cabo de medio mes se introducen los purés de frutas. Hasta 1 año, el volumen de jugo y puré consumido por día aumenta a 30-50 ml. El primer puré de verduras en agua (excepto legumbres) se introduce a los 5 meses. A partir de los 5,5 meses, a un niño se le pueden dar cereales comerciales sin lácteos (trigo sarraceno, arroz, maíz). El bebé puede probar los productos cárnicos a partir de los 6 meses, se debe dar preferencia a los alimentos enlatados especializados que no contengan leche ni productos que contengan leche.
A la hora de elegir productos de alimentación especializados para bebés, es necesario leer atentamente las etiquetas. Los productos que contienen no más de 5 mg de galactosa por 100 g se consideran seguros para un bebé. Si hay más de 20 mg de galactosa, está estrictamente prohibido darle el producto a un niño.
La idoneidad del tratamiento de la galactosemia se comprueba mediante un examen de control una vez por trimestre. También se prueba la eficacia del tratamiento de los síntomas de patología acompañantes.
¡Importante! Se presta especial atención a la selección de medicamentos para estos pacientes. Algunos medicamentos contienen lactosa, mientras que otros pueden inhibir la eliminación de productos metabólicos del hígado, lo que generalmente agrava los síntomas de una anomalía congénita y es inaceptable en el marco de un tratamiento competente.Prevención de la galactosemia

Aún no se han desarrollado medidas preventivas especializadas para la enfermedad. Si se ha descubierto que los padres tienen predisposición a la patología, se recomienda planificar la concepción con consultas adicionales con un genetista. Durante el embarazo, también debe someterse a una serie de exámenes (biopsia de vellosidades coriónicas a las 10-12 semanas y prueba de líquido amniótico a las 15-18 semanas) para detectar mutaciones en los genes GALT y GALE.
A pesar del tratamiento sintomático bastante eficaz de la patología, las perspectivas a largo plazo para el desarrollo del niño son difíciles de predecir. Por lo tanto, los indicadores de desarrollo físico del paciente serán más bajos que los de sus compañeros, es probable que se desarrollen trastornos del habla y de la coordinación, aumento de la fragilidad ósea y, en las niñas, disfunción ovárica.
A partir de los 5 años, se puede recomendar al niño que tome vitaminas y medicamentos que contengan ATP para prevenir la progresión de la enfermedad. A partir de los 12 años, a las niñas se les prescribe terapia hormonal para compensar la disfunción ovárica.
La galactosemia es una enfermedad genéticamente determinada bastante rara. El mecanismo de desarrollo de la patología aún no se ha estudiado en profundidad, pero la gravedad del problema ha llevado al desarrollo de una serie de técnicas de diagnóstico eficaces. El cribado neonatal, obligatorio en varios países, permite detectar la enfermedad a tiempo y tomar medidas. Y aunque el tratamiento de enfermedades genéticamente determinadas aún no está completamente desarrollado, la terapia ayuda a eliminar los síntomas graves de la enfermedad y su progresión. La tarea de los padres en este caso es diagnosticar la patología lo antes posible y seguir las instrucciones del médico, ayudando así al niño a afrontar la anomalía congénita.
¿Qué es la galactosemia? Mire el video:
