
Vad är galaktosemi och varför uppstår det hos ett barn? Symtom, diagnos av sjukdomen. Behandling av galaktosemi hos nyfödda och tillgängliga förebyggande åtgärder.
Innehållet i artikeln:- Vad är galaktosemi
- Utvecklingssymptom
- Diagnostik
- Hur man behandlar galaktosemi
- Förebyggande
Galaktosemi är en sällsynt men fortfarande vanlig ärftlig abnormitet i samband med metabolism. Enzymopati är förknippad med dysfunktion av kolhydratmetabolism, som ett resultat av vilket kroppen ackumulerar galaktos och dess derivat. För stora mängder galaktos leder till utvecklingen av en klinisk bild. Vid sen diagnos av galaktosemi blir symtomen uttalade och prognosen för normalisering av patientens tillstånd är dyster. Par med en predisposition för sjukdomen behöver veta om typerna av galaktosemi, metoder för diagnos, behandling och förebyggande, och också tänka på familjeplanering. Åtgärder som vidtas i rätt tid minskar risken för komplikationer hos den nyfödda avsevärt.
Vad är galaktosemi?
Innan vi pratar om rekommendationer för galaktosemi är det nödvändigt att förstå galaktosens roll i kolhydratmetabolismen. Detta enkla socker kommer in i människokroppen som en del av laktos, en disackarid vars andra komponent är glukos. Normalt används monosackariden som "energi"-resurser för celler och komplexa föreningar, "byggande" material av nervvävnad, membran och nervändar.
Hos en frisk person bryts laktos ner till dess komponenter i matsmältningskanalen och bearbetas gradvis. Således, i det första steget av bearbetningen, erhålls galaktos 1-fosfat under verkan av ett speciellt enzym galaktokinas och ATP. I nästa steg, som ett resultat av utbyte under inverkan av enzymer, erhålls glukos 1-fosfat och uridinfosfatgalaktos. Monosackarider absorberas mycket lättare av tarmarna under matsmältningen, vilket är mycket viktigt för nyfödda, eftersom mjölk är basen i deras kost.
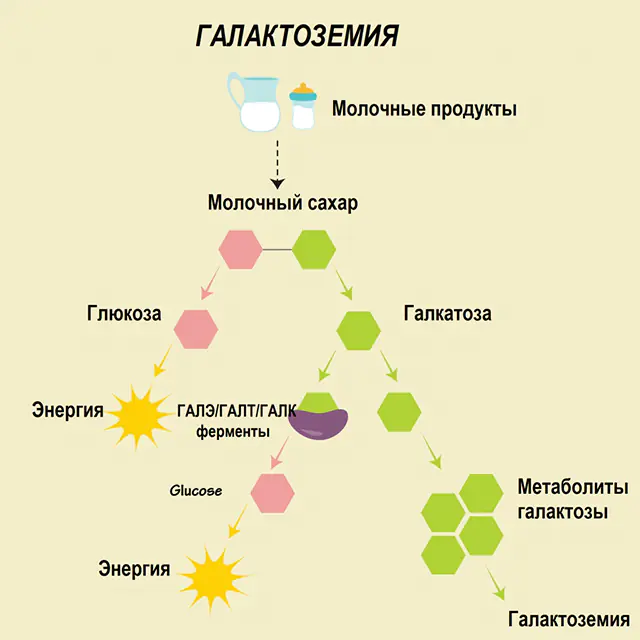
Tre enzymer är involverade i bearbetningsprocesserna - GALA (galaktokinas), GALT (galaktos 1-fosfat uridiyl transferas), GALE (epimeras). Bristfällig produktion av minst ett av dessa enzymer i det första eller andra stadiet av kolhydratmetabolism leder till ackumulering av metaboliter, förgiftning av kroppen och den efterföljande manifestationen av symtom på galaktosemi.
Beroende på det defekta enzymet särskiljs motsvarande typer av galaktosemi:
- GALT överträdelse- den första typen förekommer i ett fall per 40 tusen nyfödda; analys för galaktosemi kommer att visa minskad enzymaktivitet;
- GALC patologi- den andra typen förekommer hos ett barn av 500 tusen, medan aktiviteten av det grundläggande enzymet i erytrocyter är inom det normala intervallet, men symtomen på sjukdomen är uttalade;
- STORM— Den tredje typen registrerades minst (ett fall per miljon människor) och kännetecknas av milda symtom.
Den vanligaste typen av sjukdom (GALT) delas in i former, beroende på genetiken för galaktosemi:
- Misslyckandet är karakteristiskt för stadiet av omvandling av asparaginsyraamid till aspartat - patologin kallas också Duartes tecken (Duarte). Det är anmärkningsvärt att en person med en sådan anomali kommer att känna sig helt frisk, även om aktiviteten av GALT-enzymet minskar med 25% -50%.
- Misslyckande av glutamin-arginin-övergången registreras oftast hos kaukasier; enzymaktiviteten är bara 10% av normen, vilket provocerar ett allvarligt sjukdomsförlopp.
- En störning av arginin-tryptofanövergången uppstår när enzymet är helt inaktivt, en extremt allvarlig form av sjukdomen.
- Avbrott i lysin-asparagintransformationsprocessen inträffar ganska ofta.
- Misslyckande med serin-leucinfermentering registrerades endast hos representanter för den negroida rasen och är associerad med otillräcklig enzymaktivitet i levern (på en nivå av högst 10% av den allmänt accepterade normen).
GALT-enzymet finns i kroppen i alltför stora mängder, så en liten minskning av dess aktivitet visar sig inte alltid som kliniska symtom. Först när GALT-aktiviteten sjunker med 50 procent eller mer kommer de första tecknen på sjukdomen att visas.
Förgiftning av kroppen med galaktos och metaboliter är farligt för utvecklingen av komplikationer på lång sikt: nedsatt motorisk funktion, tal, reproduktionsfunktioner, särskilt hos flickor (trots att frekvensen av sjukdomar hos pojkar och flickor är densamma), tillväxthämning och andra utvecklingsstörningar.
Notera! Sjukdomens patogenes har ännu inte studerats fullt ut. Det har dock redan fastställts att ackumulering av galaktos i kroppen är farlig inte bara på grund av dess toxiska dosering, utan också på grund av dess hämmande effekt på andra enzymer. Utan korrekt behandling och kontroll kan patienten utveckla hypoglykemiskt syndrom.Som redan nämnts är patologin en medfödd anomali. Typen av arv av galaktosemi är autosomal recessiv, det vill säga barnet får defekta gener från föräldrarna. Den muterade genen kan hittas i olika alleler (former av tillståndet) - defekta och normala, när defekten överfördes från endast en förälder. I detta fall manifesterar sjukdomen sig i mindre utsträckning. Men om defekta alleler har överförts från både fadern och modern i enlighet med typen av arv av galaktosemi, kommer de första tecknen på sjukdomen att uppträda inom några dagar efter barnets födelse. Det är anmärkningsvärt att om en sjukdom diagnostiseras hos ett barn i en familj, ökar sannolikheten för att patologin inträffar hos nästa barn från samma föräldrar med 25%.
Otillräcklig produktion av enzymer leder till ackumulering av galaktos i kroppen, och som ett resultat uppträder tecken på galaktosemi och utsöndringssystemets funktioner störs. Utan behandling inom några månader av livet dör en person av leversvikt eller infektioner som påverkar kroppen.
Det är viktigt att huvudorsakerna till galaktosemi (ärftlig komponent) kan fastställas redan före barnets födelse, men läkare utesluter inte påverkan av andra indikatorer på utvecklingen av patologin:
- andra genmutationer;
- påverkad vävnad i levern och centrala nervsystemet;
- ansamling av vätska i ögats lins, dimsyn.
Och ändå, om en patient får diagnosen galaktosemi, spelar genetik en nyckelroll. Samtidiga faktorer är viktiga endast hos heterozygota patienter, det vill säga de hos vilka den defekta genen överfördes från endast en förälder. Hos homozygota patienter är själva patologin allvarlig och kräver högspecialiserad behandling.
Symtom på utvecklingen av galaktosemi
Beroende på svårighetsgraden av manifestationerna av tecken på galaktosemi särskiljs tre grader av sjukdomen:
- ljus- upptäcks vanligtvis av misstag på grund av mjölkintolerans;
- genomsnitt- de första tecknen uppträder först efter att ha druckit mjölk;
- tung— symtomen på patologin överlagras på andra störningar i kroppen (vätskeansamling i bukhålan, sepsis).
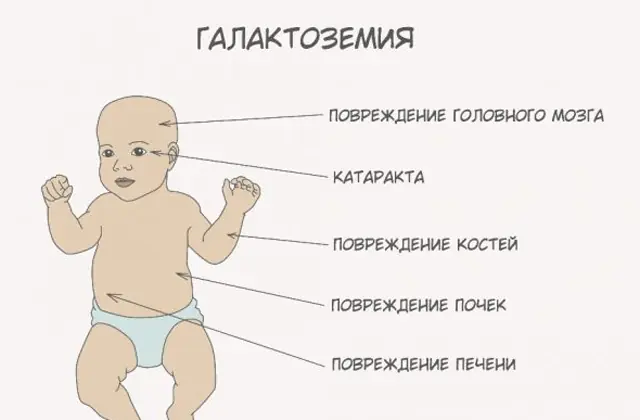
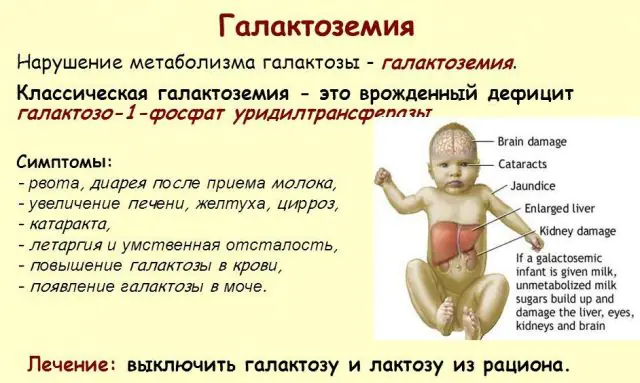
Klassisk GALT-patologi manifesterar sig i sin mest allvarliga form. De första tecknen på sjukdomen kommer att bli synliga efter barnets födelse och hans första matning, eftersom mjölk utgör den nyföddas huvuddiet. Efter matning upplever han kräkningar, frekventa tarmrörelser och ökad dåsighet mot bakgrund av konstant slöhet och muskelhypotoni. Trots regelbunden amning sker ingen viktökning. Matsmältningsstörningar är ett av de första tecknen på dysfunktion.
När kroppen blir berusad blir tecken på leverskador uttalade - gulsot, förstoring av levern. Inom några veckor utvecklar barnet grå starr, och efter några månader, på grund av förgiftning av nervvävnaderna, noteras störningar i psykomotoriska funktioner. Njurtubuliernas aktivitet störs, som ett resultat av vilket minskande socker uppträder i urinen och minskad blodkoagulering noteras.
Utan den nödvändiga terapin förvärras symtomen när barnets kropp blir berusad, och risken för normal utveckling och till och med mänskligt liv ökar. 20-30% av barnen, om behandling avvisas eller sjukdomen diagnostiseras sent, dör av sepsis, som utvecklas på grund av leukocytotisk aktivitet. De som överlever lider av njursvikt och problem med psykomotorisk utveckling.
På många sätt beror graden av manifestation av patologin på den ärftliga orsaken. Galaktosemi visar sig när GALT-brist uppstår. Således kan Duharts tecken till en början bara avslöja sig som långvarig gulsot (upp till 2 månader efter födseln), grå starr och leverdysfunktion diagnostiseras lite senare. Men om aktiviteten av GALT-enzymet är minst 50 % kan det hända att kliniska symtom inte uppträder.
Bristen på GALA-enzym är inte så uttalad. Det huvudsakliga symtomet på sjukdomen är progressiv grå starr, och monosackarider kan också förekomma i urinen med tiden. Samtidigt är barnets viktminskning obetydlig, och psykomotoriska färdigheter utvecklas inom det normala området.
GALE-brist är initialt extremt sällsynt, men symtomen delas fortfarande upp i två former: godartad, när bristen på enzymet påverkar blodkropparna, och generaliserad, när bristen påverkar alla vävnader i kroppen. I det första fallet kommer endast en analys för galaktosemi att hjälpa till att upptäcka sjukdomen i de tidiga stadierna; först med tiden kommer dysfunktion av psykomotoriska färdigheter och talutveckling att noteras. Den andra formen liknar i sina symtom GALT-brist, men i detta fall finns det också en förstoring av mjälten. Även vid behandling av en allvarlig form av GALE-sjukdom upplever barnet med tiden försenad psykomotorisk utveckling, dövhet och synproblem.
Diagnos av galaktosemi
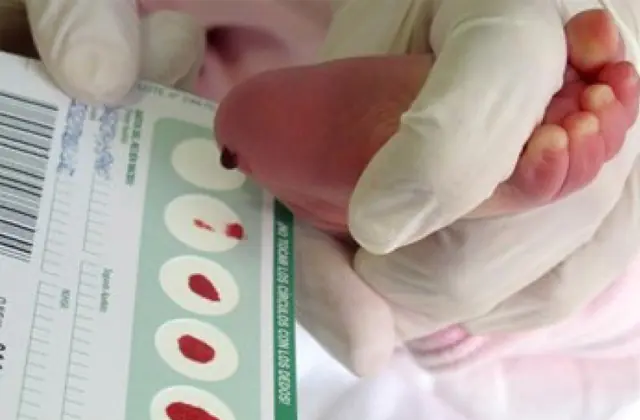
Neonatal screening, införd i ett antal länder, gör det möjligt att identifiera upp till 50 medfödda sjukdomar i tidiga skeden, inklusive otillräcklig produktion av enzymer som är aktivt involverade i ämnesomsättningen. Om screening av någon anledning inte utförs identifieras sjukdomen utifrån aggregerade egenskaper.
Så om galaktosemi misstänks, görs läkarens rekommendationer först efter en detaljerad diagnos. En liten patient måste genomgå:
- allmän undersökning - om tecken på gulsot finns;
- allmänna tecken bedöms - kvantiteten och kvaliteten på tarmrörelser, närvaron eller frånvaron av muskelhypotoni, problem med psykomotoriska färdigheter;
- laboratorietester - blodprov för biokemi, allmänt blodprov, urinprov för socker och protein;
- ytterligare undersökning av leverns och njurarnas tillstånd.
DNA-diagnostik är obligatoriskt för att upptäcka GALT-mutationer. Om barnet uppvisar symtom på sjukdomen, och aktiviteten av GALT-enzymer enligt testresultat är normal, utförs DNA-diagnostik för GALE-enzymet.
Läkare kan ordinera ett antal ytterligare tester för att differentiera diagnosen. Exklusiv diagnos är nödvändig för att bekräfta eller motbevisa närvaron av diabetes mellitus och andra patologier associerade med höga sockernivåer, såväl som sjukdomar som orsakar leverförstoring.
Hur behandlar man galaktosemi?

När neonatal screening och andra undersökningar har bekräftat förekomsten av galaktosemi, ordineras behandling. Det är nödvändigt att förstå att eftersom sjukdomen orsakas av en genetisk predisposition, är ett fullständigt botemedel i detta stadium av medicinsk utveckling omöjligt. Terapi syftar till att minska de kliniska manifestationerna av patologin, minimera utvecklingen av sjukdomen och utvecklingen av komplikationer.
Den huvudsakliga behandlingsmetoden är rätt näring för galaktosemi. Alla mjölkhaltiga produkter, även bröd, godis, korv och andra, är helt uteslutna från patientens kost. Produkter av vegetabiliskt och animaliskt ursprung som innehåller oligosackarider tas också bort: baljväxter, soja, spenat, choklad, nötter, lever, hjärnor, ägg och derivat av dessa produkter. Strikt kostterapi är det enda sättet att förhindra förgiftning av kroppen.
För nyfödda är näring för galaktosemi också specialiserad. Bröstmjölk elimineras gradvis helt från kosten och ersätts med laktosfria formuleringar och formuleringar baserade på sojaproteinisolat. Volymen av näringsingredienser bör i detta fall motsvara standarderna för ett friskt barn. Om ett barn uppvisar symptom på allergi mot sojaproteinisolat, väljs blandningar baserade på kaseinhydrolysat. Det första intaget av matblandning bör vara 1/10 av barnets dagliga näringsbehov. Inom 5-7 dagar elimineras bröstmjölken helt från kosten.
De första kompletterande livsmedel för barn med patologi ordineras från 4 månader. Dieten utökas med fruktjuicer (äpple, päron och andra). Efter en halv månad introduceras fruktpuréer. Upp till 1 år ökar volymen juice och puré som konsumeras per dag till 30-50 ml. Den första grönsakspurén i vatten (förutom baljväxter) introduceras vid 5 månader. Från 5,5 månader kan ett barn ges mejerifria kommersiella spannmål (bovete, ris, majs). Barnet kan prova köttprodukter från 6 månaders ålder; specialmat som inte innehåller mjölk eller mjölkhaltiga produkter bör föredras.
När du väljer specialiserade livsmedelsprodukter för spädbarn måste du noggrant läsa etiketterna. Produkter som inte innehåller mer än 5 mg galaktos per 100 g anses vara säkra för ett barn. Om det finns mer än 20 mg galaktos är det strängt förbjudet att ge produkten till ett barn.
Behandlingens tillräcklighet för galaktosemi kontrolleras genom en kontrollundersökning en gång i kvartalet. Effektiviteten av behandlingen av åtföljande symtom på patologi testas också.
Viktig! Särskild uppmärksamhet ägnas åt valet av mediciner för sådana patienter. Vissa läkemedel innehåller laktos i sin sammansättning, medan andra kan hämma avlägsnandet av metaboliska produkter från levern, vilket i allmänhet förvärrar symtomen på den medfödda anomalien och är oacceptabel inom ramen för kompetent behandling.Förebyggande av galaktosemi

Specialiserade förebyggande åtgärder för sjukdomen har ännu inte utvecklats. Om föräldrar har visat sig ha en anlag för patologin, rekommenderas att planera befruktningen med ytterligare konsultationer med en genetiker. Under graviditeten bör du också genomgå en rad undersökningar (chorionic villus biopsi vid 10-12 veckor och fostervattenprov vid 15-18 veckor) för att upptäcka GALT och GALE mutationer i gener.
Trots den utvecklade ganska effektiva symtomatiska behandlingen av patologin är de långsiktiga utsikterna för barnets utveckling svåra att förutsäga. Således kommer patientens fysiska utvecklingsindikatorer att vara lägre än hos jämnåriga, tal- och koordinationsstörningar kommer sannolikt att utvecklas, ökad benskörhet och hos flickor - äggstocksdysfunktion.
Från 5 års ålder kan ett barn rekommenderas att ta vitaminer och läkemedel som innehåller ATP för att förhindra utvecklingen av sjukdomen. Från 12 års ålder ordineras flickor hormonbehandling för att kompensera för äggstocksdysfunktion.
Galaktosemi är en ganska sällsynt genetiskt betingad sjukdom. Mekanismen för utveckling av patologin har ännu inte studerats noggrant, men problemets allvar har lett till utvecklingen av ett antal effektiva diagnostiska tekniker. Neonatal screening, obligatorisk i ett antal länder, gör det möjligt att upptäcka sjukdomen i tid och vidta åtgärder. Och även om behandlingen av genetiskt betingade sjukdomar inte har utvecklats fullt ut hittills, hjälper terapi att bli av med allvarliga symtom på sjukdomen och dess framsteg. Föräldrarnas uppgift i det här fallet är att diagnostisera patologin så tidigt som möjligt och följa läkarens instruktioner, vilket hjälper barnet att hantera den medfödda anomalien.
Vad är galaktosemi - se videon:
