
Qu'est-ce que la galactosémie et pourquoi survient-elle chez un enfant ? Symptômes, diagnostic de la maladie. Traitement de la galactosémie chez les nouveau-nés et mesures préventives disponibles.
Le contenu de l'article :- Qu'est-ce que la galactosémie
- Symptômes de développement
- Diagnostique
- Comment traiter la galactosémie
- La prévention
La galactosémie est une anomalie héréditaire rare mais encore courante associée au métabolisme. L'enzymopathie est associée à un dysfonctionnement du métabolisme des glucides, à la suite duquel le corps accumule du galactose et ses dérivés. Des quantités excessives de galactose conduisent au développement d'un tableau clinique. En cas de diagnostic tardif de galactosémie, les symptômes deviennent prononcés et le pronostic de normalisation de l’état du patient est sombre. Les couples prédisposés à la maladie doivent connaître les types de galactosémie, les méthodes de diagnostic, de traitement et de prévention, et également réfléchir à la planification familiale. Des mesures prises en temps opportun réduisent considérablement le risque de complications chez le nouveau-né.
Qu’est-ce que la galactosémie ?
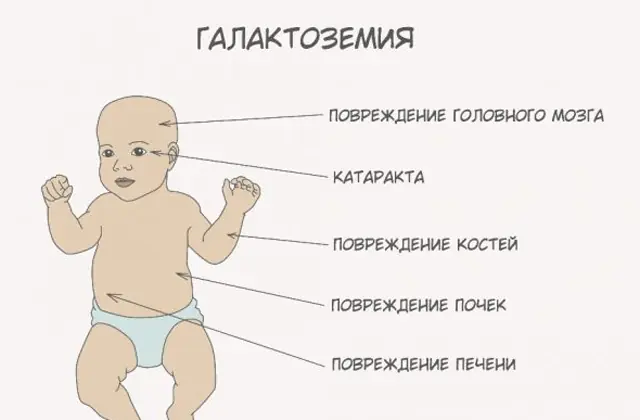
Avant de parler de recommandations concernant la galactosémie, il est nécessaire de comprendre le rôle du galactose dans le métabolisme des glucides. Ce sucre simple pénètre dans le corps humain sous forme de lactose, un disaccharide dont le deuxième composant est le glucose. Normalement, le monosaccharide est utilisé comme ressource « énergétique » pour les cellules et les composés complexes, matériaux de « construction » du tissu nerveux, des membranes et des terminaisons nerveuses.
Chez une personne en bonne santé, le lactose est décomposé en ses composants dans le tube digestif et progressivement transformé. Ainsi, lors de la première étape du traitement, le galactose 1-phosphate est obtenu sous l'action d'une enzyme spéciale, la galactokinase et l'ATP. À l'étape suivante, à la suite d'un échange sous l'action d'enzymes, on obtient du glucose 1-phosphate et du phosphate d'uridine galactose. Les monosaccharides sont beaucoup plus facilement absorbés par les intestins lors de la digestion, ce qui est très important pour les nouveau-nés, puisque le lait constitue la base de leur alimentation.
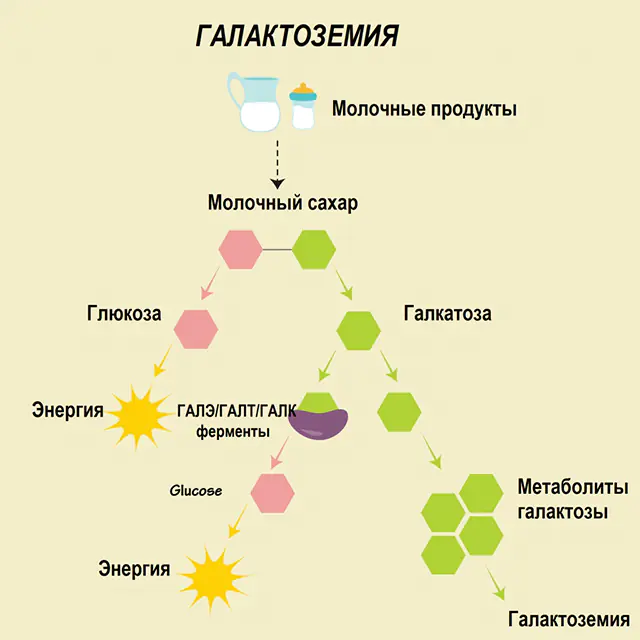
Trois enzymes sont impliquées dans les processus de traitement - GALA (galactokinase), GALT (galactose 1-phosphate uridiyl transférase), GALE (épimérase). Une production déficiente d'au moins une de ces enzymes au premier ou au deuxième stade du métabolisme des glucides entraîne l'accumulation de métabolites, une intoxication du corps et la manifestation ultérieure de symptômes de galactosémie.
Selon l'enzyme défectueuse, on distingue les types correspondants de galactosémie :
- Violation de la loi GALT- le premier type survient dans un cas sur 40 000 nouveau-nés ; l'analyse de la galactosémie montrera une activité enzymatique réduite ;
- Pathologie GALC- le deuxième type survient chez un enfant sur 500 000, alors que l'activité de l'enzyme basique dans les érythrocytes se situe dans la plage normale, mais les symptômes de la maladie sont prononcés ;
- GRAND VENT— le troisième type est celui qui a été le moins enregistré (un cas par million de personnes) et se caractérise par des symptômes légers.
Le type de maladie le plus courant (GALT) est divisé en formes, selon la génétique de la galactosémie :
- L'échec est caractéristique du stade de transformation de l'amide d'acide aspartique en aspartate - la pathologie est également appelée signe de Duarte (Duarte). Il est à noter qu'une personne présentant une telle anomalie se sentira en parfaite santé, bien que l'activité de l'enzyme GALT soit réduite de 25 à 50 %.
- L'échec de la transition glutamine-arginine est le plus souvent enregistré chez les Caucasiens, l'activité enzymatique n'est que de 10 % de la norme, ce qui provoque une évolution sévère de la maladie.
- Une perturbation de la transition arginine-tryptophane se produit lorsque l’enzyme est totalement inactive, une forme extrêmement grave de la maladie.
- La perturbation du processus de transformation lysine-asparagine se produit assez souvent.
- L'échec de la fermentation sérine-leucine n'a été enregistré que chez les représentants de la race négroïde et est associé à une activité enzymatique insuffisante dans le foie (à un niveau ne dépassant pas 10 % de la norme généralement acceptée).
L'enzyme GALT est présente dans l'organisme en quantités excessives, de sorte qu'une légère diminution de son activité ne se manifeste pas toujours par des symptômes cliniques. Ce n’est que lorsque l’activité du GALT chute de 50 pour cent ou plus que les premiers signes de la maladie apparaissent.
L'intoxication de l'organisme par le galactose et les métabolites est dangereuse pour le développement de complications à long terme : troubles de la motricité, de la parole, des fonctions reproductives, notamment chez les filles (malgré le fait que la fréquence des maladies chez les garçons et les filles soit la même), retard de croissance et autres dysfonctionnements du développement.
Note! La pathogenèse de la maladie n'a pas encore été entièrement étudiée. Cependant, il a déjà été établi que l'accumulation de galactose dans l'organisme est dangereuse non seulement en raison de son dosage toxique, mais également en raison de son effet inhibiteur sur d'autres enzymes. Sans traitement et contrôle appropriés, le patient peut développer un syndrome hypoglycémique.Comme déjà indiqué, la pathologie est une anomalie congénitale. Le type de transmission de la galactosémie est autosomique récessif, c'est-à-dire que l'enfant reçoit des gènes défectueux des parents. Le gène muté peut être trouvé dans différents allèles (formes de la maladie) - défectueux et normaux, lorsque le défaut a été transmis par un seul parent. Dans ce cas, la maladie se manifeste dans une moindre mesure. Mais si des allèles défectueux ont été transmis à la fois par le père et par la mère conformément au type d'hérédité de la galactosémie, les premiers signes de la maladie apparaîtront quelques jours après la naissance du bébé. Il est à noter que si une maladie est diagnostiquée chez un enfant d'une famille, la probabilité que la pathologie survienne chez l'enfant suivant des mêmes parents augmente de 25 %.
Une production insuffisante d'enzymes entraîne une accumulation de galactose dans l'organisme et, par conséquent, des signes de galactosémie apparaissent et les fonctions du système excréteur sont perturbées. Sans traitement, après quelques mois de vie, une personne meurt d’une insuffisance hépatique ou d’infections affectant le corps.
Il est important que les principales causes de la galactosémie (composante héréditaire) puissent être établies avant même la naissance de l'enfant, mais les médecins n'excluent pas l'influence d'autres indicateurs sur le développement de la pathologie :
- d'autres mutations génétiques ;
- tissus affectés du foie et du système nerveux central ;
- accumulation de liquide dans le cristallin de l’œil, vision floue.
Et pourtant, si un patient reçoit un diagnostic de galactosémie, la génétique joue un rôle clé. Les facteurs concomitants ne sont importants que chez les patients hétérozygotes, c'est-à-dire ceux chez qui le gène défectueux a été transmis par un seul parent. Chez les patients homozygotes, la pathologie elle-même est sévère et nécessite un traitement hautement spécialisé.
Symptômes du développement de la galactosémie
Selon la gravité des manifestations des signes de galactosémie, on distingue trois degrés de la maladie :
- lumière- généralement détecté accidentellement en raison d'une intolérance au lait ;
- moyenne- les premiers signes n'apparaissent qu'après avoir bu du lait ;
- lourd— les symptômes de la pathologie se superposent à d'autres dysfonctionnements de l'organisme (accumulation de liquide dans la cavité abdominale, septicémie).
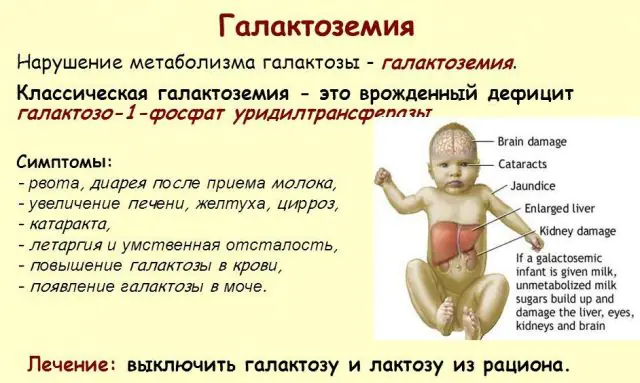
La pathologie classique du GALT se manifeste sous sa forme la plus sévère. Les premiers signes de la maladie deviendront visibles après la naissance de l'enfant et sa première tétée, puisque le lait constitue l'alimentation principale du nouveau-né. Après s'être nourri, il éprouve des vomissements, des selles fréquentes et une somnolence accrue dans le contexte d'une léthargie constante et d'une hypotension musculaire. Malgré un allaitement régulier, il n’y a pas de prise de poids. Les troubles digestifs sont l’un des premiers signes de dysfonctionnement.
À mesure que le corps devient intoxiqué, les signes de lésions hépatiques deviennent prononcés - jaunisse, hypertrophie du foie. En quelques semaines, l'enfant développe des cataractes, et après quelques mois, en raison d'une intoxication des tissus nerveux, on note des troubles des fonctions psychomotrices. L'activité des tubules rénaux est perturbée, ce qui entraîne l'apparition de sucres réducteurs dans l'urine et une diminution de la coagulation sanguine.
Sans le traitement nécessaire, les symptômes s’aggravent à mesure que le corps du bébé devient intoxiqué et le risque pour son développement normal, voire pour sa vie humaine, augmente. 20 à 30 % des enfants, si le traitement est refusé ou si la maladie est diagnostiquée tardivement, meurent d'une septicémie, qui se développe en raison de l'activité leucocytose. Ceux qui survivent souffrent d’insuffisance rénale et de problèmes de développement psychomoteur.
À bien des égards, le degré de manifestation de la pathologie dépend de la cause héréditaire. La galactosémie se manifeste par un déficit en GALT. Ainsi, le signe de Duhart ne peut se révéler dans un premier temps que par un ictère prolongé (jusqu'à 2 mois après la naissance), des cataractes et des dysfonctionnements hépatiques sont diagnostiqués un peu plus tard. Mais si l'activité de l'enzyme GALT est d'au moins 50 %, les symptômes cliniques peuvent ne pas apparaître.
Le manque d'enzyme GALA n'est pas si prononcé. Le principal symptôme de la maladie est la cataracte progressive et des monosaccharides peuvent également apparaître dans l'urine au fil du temps. Dans le même temps, la perte de poids du bébé est insignifiante et les capacités psychomotrices se développent dans les limites normales.
Le déficit en GALE est initialement extrêmement rare, mais les symptômes sont encore divisés en deux formes : bénin, lorsque le manque de l'enzyme affecte les cellules sanguines, et généralisé, lorsque le déficit affecte tous les tissus du corps. Dans le premier cas, seule une analyse de la galactosémie permettra de détecter la maladie à un stade précoce, ce n'est qu'avec le temps que l'on constatera un dysfonctionnement des capacités psychomotrices et du développement de la parole. La deuxième forme présente des symptômes similaires à ceux du déficit en GALT, mais dans ce cas, il existe également une hypertrophie de la rate. Même avec le traitement d'une forme grave de la maladie GALE, l'enfant présente au fil du temps un retard de développement psychomoteur, une surdité et des problèmes de vision.
Diagnostic de la galactosémie
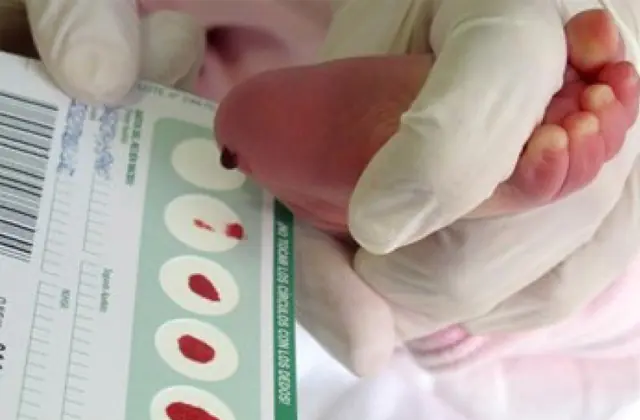
Le dépistage néonatal, introduit dans plusieurs pays, permet d'identifier à un stade précoce jusqu'à 50 maladies congénitales, dont une production insuffisante d'enzymes activement impliquées dans le métabolisme. Si, pour une raison quelconque, le dépistage n'est pas effectué, la maladie est identifiée sur la base de caractéristiques globales.
Ainsi, si une galactosémie est suspectée, les recommandations du médecin ne sont faites qu’après un diagnostic détaillé. Un petit patient doit subir :
- examen général - présence de signes de jaunisse ;
- les signes généraux sont évalués - la quantité et la qualité des selles, la présence ou l'absence d'hypotonie musculaire, les problèmes de psychomotricité ;
- tests de laboratoire - test sanguin pour la biochimie, test sanguin général, test urinaire pour le sucre et les protéines ;
- examen complémentaire de l'état du foie et des reins.
Les diagnostics ADN sont obligatoires pour détecter les mutations GALT. Si l'enfant présente des symptômes de la maladie et que l'activité des enzymes GALT selon les résultats des tests est normale, un diagnostic ADN est effectué pour l'enzyme GALE.
Les médecins peuvent prescrire un certain nombre de tests supplémentaires pour différencier le diagnostic. Un diagnostic exclusif est nécessaire pour confirmer ou infirmer la présence de diabète sucré et d'autres pathologies associées à des taux de sucre élevés, ainsi que des maladies provoquant une hypertrophie du foie.
Comment traiter la galactosémie ?

Une fois que le dépistage néonatal et d’autres examens ont confirmé la présence d’une galactosémie, un traitement est prescrit. Il faut comprendre que puisque la maladie est causée par une prédisposition génétique, une guérison complète à ce stade du développement médical est impossible. La thérapie vise à réduire les manifestations cliniques de la pathologie, en minimisant la progression de la maladie et le développement de complications.
La principale méthode de traitement est une bonne nutrition pour la galactosémie. Tous les produits contenant du lait, même le pain, les sucreries, les saucisses et autres, sont totalement exclus de l'alimentation du patient. Les produits d'origine végétale et animale contenant des oligosaccharides sont également supprimés : les légumineuses, le soja, les épinards, le chocolat, les fruits à coque, le foie, la cervelle, les œufs et les dérivés de ces produits. Une thérapie diététique stricte est le seul moyen de prévenir l'intoxication du corps.
Pour les nouveau-nés, la nutrition pour la galactosémie est également spécialisée. Le lait maternel est progressivement complètement éliminé de l'alimentation et remplacé par des préparations sans lactose et à base d'isolat de protéines de soja. Dans ce cas, le volume d'ingrédients nutritionnels doit correspondre aux normes pour un enfant en bonne santé. Si un bébé présente des symptômes d'allergie à l'isolat de protéines de soja, des mélanges à base d'hydrolysats de caséine sont sélectionnés. La première prise de mélange alimentaire doit représenter 1/10 des besoins nutritionnels quotidiens de l’enfant. En 5 à 7 jours, le lait maternel est complètement éliminé de l'alimentation.
Les premiers aliments complémentaires pour les enfants atteints de pathologie sont prescrits à partir de 4 mois. L'alimentation est complétée par des jus de fruits (pomme, poire et autres). Au bout d'un demi-mois, les purées de fruits sont introduites. Jusqu'à 1 an, le volume de jus et de purée consommé par jour augmente jusqu'à 30-50 ml. La première purée de légumes dans l'eau (sauf les légumineuses) est introduite à 5 mois. A partir de 5,5 mois, un enfant peut recevoir des céréales commerciales sans produits laitiers (sarrasin, riz, maïs). Le bébé peut essayer les produits carnés dès l'âge de 6 mois, la préférence doit être donnée aux aliments spécialisés en conserve qui ne contiennent pas de lait ni de produits contenant du lait.
Lorsque vous choisissez des produits alimentaires spécialisés pour nourrissons, vous devez lire attentivement les étiquettes. Les produits qui ne contiennent pas plus de 5 mg de galactose pour 100 g sont considérés comme sans danger pour un bébé. S'il y a plus de 20 mg de galactose, il est strictement interdit de donner le produit à un enfant.
L'adéquation du traitement de la galactosémie est vérifiée par un examen de contrôle une fois par trimestre. L'efficacité du traitement des symptômes accompagnant la pathologie est également testée.
Important! Une attention particulière est accordée à la sélection des médicaments pour ces patients. Certains médicaments contiennent du lactose dans leur composition, tandis que d'autres peuvent inhiber l'élimination des produits métaboliques du foie, ce qui aggrave généralement les symptômes de l'anomalie congénitale et est inacceptable dans le cadre d'un traitement compétent.Prévention de la galactosémie

Des mesures préventives spécialisées contre la maladie n'ont pas encore été développées. S'il s'avère que les parents ont une prédisposition à la pathologie, il est recommandé de planifier la conception avec des consultations supplémentaires avec un généticien. Pendant la grossesse, vous devez également subir une série d'examens (biopsie des villosités choriales à 10 à 12 semaines et analyse du liquide amniotique à 15 à 18 semaines) pour détecter les mutations GALT et GALE dans les gènes.
Malgré le traitement symptomatique assez efficace de la pathologie, les perspectives à long terme du développement de l’enfant sont difficiles à prédire. Ainsi, les indicateurs de développement physique de la patiente seront inférieurs à ceux de ses pairs, des troubles de la parole et de la coordination, une fragilité osseuse accrue et chez les filles un dysfonctionnement ovarien risquent de se développer.
Dès l'âge de 5 ans, il peut être conseillé à un enfant de prendre des vitamines et des médicaments contenant de l'ATP pour prévenir la progression de la maladie. Dès l'âge de 12 ans, les filles se voient prescrire un traitement hormonal pour compenser le dysfonctionnement ovarien.
La galactosémie est une maladie génétiquement déterminée assez rare. Le mécanisme de développement de la pathologie n'a pas encore été étudié de manière approfondie, mais la gravité du problème a conduit au développement d'un certain nombre de techniques de diagnostic efficaces. Le dépistage néonatal, obligatoire dans de nombreux pays, permet de détecter la maladie à temps et d'agir. Et bien que le traitement des maladies génétiquement déterminées ne soit pas encore complètement développé, la thérapie aide à se débarrasser des symptômes graves de la maladie et de sa progression. La tâche des parents dans ce cas est de diagnostiquer la pathologie le plus tôt possible et de suivre les instructions du médecin, aidant ainsi l'enfant à faire face à l'anomalie congénitale.
Qu'est-ce que la galactosémie - regardez la vidéo :
