
Qalaktozemiya nədir və niyə uşaqda baş verir? Xəstəliyin simptomları, diaqnozu. Yenidoğulmuşlarda qalaktozemiyanın müalicəsi və mövcud profilaktik tədbirlər.
Məqalənin məzmunu:- Qalaktozemiya nədir
- İnkişaf əlamətləri
- Diaqnostika
- Qalaktozemiyanı necə müalicə etmək olar
- Qarşısının alınması
Qalaktozemiya maddələr mübadiləsi ilə əlaqəli nadir, lakin hələ də ümumi irsi anormallıqdır. Enzimopatiya karbohidrat mübadiləsinin disfunksiyası ilə əlaqələndirilir, bunun nəticəsində orqanizmdə qalaktoza və onun törəmələri toplanır. Həddindən artıq miqdarda qalaktoz klinik mənzərənin inkişafına səbəb olur. Qalaktozemiyanın gec diaqnozu halında, simptomlar tələffüz olunur və xəstənin vəziyyətinin normallaşması üçün proqnoz qaranlıqdır. Xəstəliyə meylli cütlüklər qalaktozemiya növləri, diaqnostika, müalicə və profilaktika üsulları haqqında bilməli, həmçinin ailə planlaması haqqında düşünməlidirlər. Vaxtında görülən tədbirlər yenidoğanda ağırlaşma riskini əhəmiyyətli dərəcədə azaldır.
Qalaktozemiya nədir?
Qalaktozemiya üçün tövsiyələr haqqında danışmazdan əvvəl, karbohidrat mübadiləsində qalaktozanın rolunu başa düşmək lazımdır. Bu sadə şəkər insan orqanizminə ikinci komponenti qlükoza olan disakarid olan laktozanın bir hissəsi kimi daxil olur. Normalda monosaxarid hüceyrələr və kompleks birləşmələr, sinir toxumasının, membranların və sinir uclarının "tikinti" materialları üçün "enerji" mənbəyi kimi istifadə olunur.
Sağlam bir insanda laktoza həzm sistemində onun komponentlərinə parçalanır və tədricən emal olunur. Beləliklə, emalın birinci mərhələsində qalaktoza 1-fosfat xüsusi bir ferment galaktokinazın və ATP-nin təsiri altında əldə edilir. Növbəti mərhələdə fermentlərin təsiri altında mübadilə nəticəsində qlükoza 1-fosfat və uridinfosfat qalaktoza alınır. Monosakkaridlər həzm zamanı bağırsaqlar tərəfindən daha asan əmilir, bu, yenidoğulmuşlar üçün çox vacibdir, çünki süd onların pəhrizinin əsasını təşkil edir.
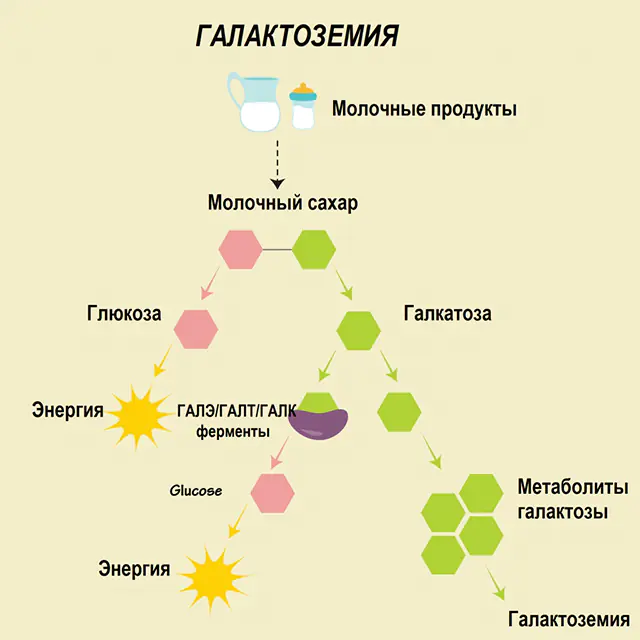
Emal proseslərində üç ferment iştirak edir - GALA (qalaktokinaz), GALT (qalaktoza 1-fosfat uridiil transferaz), GALE (epimeraz). Karbohidrat mübadiləsinin birinci və ya ikinci mərhələsində həmin fermentlərdən ən azı birinin çatışmazlığı metabolitlərin yığılmasına, orqanizmin intoksikasiyasına və sonradan qalaktozemiya əlamətlərinin təzahürünə səbəb olur.
Qüsurlu fermentdən asılı olaraq qalaktozemiyanın müvafiq növləri fərqləndirilir:
- GALT pozuntusu- birinci növ 40 min yeni doğulmuş körpəyə bir halda baş verir; qalaktozemiya üçün analiz ferment aktivliyinin azaldığını göstərəcək;
- GALC patologiyası- ikinci tip 500 min uşaqdan birində baş verir, eritrositlərdə əsas fermentin aktivliyi normal həddə olsa da, xəstəliyin əlamətləri açıq şəkildə ifadə edilir;
- GALE— üçüncü növ ən az qeydə alınıb (bir milyon nəfərə bir hal) və yüngül simptomlarla xarakterizə olunur.
Ən çox yayılmış xəstəlik növü (GALT) qalaktozemiyanın genetikasından asılı olaraq formalara bölünür:
- Uğursuzluq aspartik turşu amidinin aspartata çevrilməsi mərhələsi üçün xarakterikdir - patoloji Duarte əlaməti (Duarte) adlanır. Maraqlıdır ki, belə anomaliyaya malik olan şəxs GALT fermentinin aktivliyi 25%-50% azalsa da, özünü tamamilə sağlam hiss edəcək.
- Qlutamin-arginin keçidinin uğursuzluğu ən çox Qafqazlılarda qeyd olunur, ferment aktivliyi normanın yalnız 10% -ni təşkil edir ki, bu da xəstəliyin ağır gedişatına səbəb olur.
- Arginin-triptofan keçidinin pozulması, ferment tamamilə qeyri-aktiv olduqda baş verir, xəstəliyin son dərəcə ağır forması.
- Lizin-asparagin çevrilmə prosesinin pozulması olduqca tez-tez baş verir.
- Serin-lösin fermentasiyasının uğursuzluğu yalnız Negroid irqinin nümayəndələrində qeydə alınıb və qaraciyərdə kifayət qədər ferment fəaliyyətinin olmaması ilə əlaqələndirilir (ümumi qəbul edilmiş normanın 10% -dən çox olmayan səviyyədə).
GALT fermenti bədəndə həddindən artıq miqdarda olur, buna görə də onun fəaliyyətində bir qədər azalma həmişə klinik simptomlar kimi özünü göstərmir. Yalnız GALT aktivliyi 50 faiz və ya daha çox azaldıqda xəstəliyin ilk əlamətləri görünəcək.
Bədənin qalaktoza və metabolitlərlə intoksikasiyası uzun müddətli fəsadların inkişafı üçün təhlükəlidir: motor funksiyasının, nitqin, reproduktiv funksiyaların pozulması, xüsusən qızlarda (oğlanlarda və qızlarda xəstəliklərin tezliyi eyni olmasına baxmayaraq), böyümə geriliyi və digər inkişaf disfunksiyaları.
Qeyd! Xəstəliyin patogenezi hələ tam öyrənilməmişdir. Bununla belə, artıq müəyyən edilmişdir ki, qalaktozanın orqanizmdə toplanması təkcə onun zəhərli dozasına görə deyil, həm də digər fermentlərə inhibitor təsirinə görə təhlükəlidir. Müvafiq müalicə və nəzarət olmadan xəstədə hipoqlikemik sindrom inkişaf edə bilər.Artıq qeyd edildiyi kimi, patoloji anadangəlmə anomaliyadır. Qalaktozemiyanın irsiyyət növü autosomal resessivdir, yəni uşaq valideynlərdən qüsurlu genlər alır. Mutasiya edilmiş gen müxtəlif allellərdə (vəziyyətin formalarında) tapıla bilər - qüsurlu və normal, qüsur yalnız bir valideyndən ötürüldükdə. Bu vəziyyətdə xəstəlik daha az dərəcədə özünü göstərir. Ancaq qalaktozemiyanın irsiyyət növünə uyğun olaraq həm atadan, həm də anadan qüsurlu allellər ötürülübsə, xəstəliyin ilk əlamətləri körpənin doğulmasından bir neçə gün sonra görünəcək. Maraqlıdır ki, bir ailədə bir uşaqda xəstəlik aşkar edilərsə, eyni valideynlərdən gələn növbəti uşaqda patologiyanın baş vermə ehtimalı 25% artır.
Fermentlərin kifayət qədər istehsal olunmaması orqanizmdə qalaktozanın toplanmasına gətirib çıxarır və nəticədə qalaktozemiya əlamətləri yaranır və ifrazat sisteminin funksiyaları pozulur. Həyatdan bir neçə ay ərzində müalicə edilmədikdə, bir insan qaraciyər çatışmazlığından və ya bədənə təsir edən infeksiyalardan ölür.
Qalaktozemiyanın əsas səbəblərini (irsi komponent) uşağın doğulmasından əvvəl də müəyyən etmək vacibdir, lakin həkimlər digər göstəricilərin patologiyanın inkişafına təsirini istisna etmirlər:
- digər gen mutasiyaları;
- qaraciyərin və mərkəzi sinir sisteminin təsirlənmiş toxuması;
- gözün lensində mayenin yığılması, bulanıq görmə.
Və buna baxmayaraq, xəstəyə qalaktozemiya diaqnozu qoyularsa, genetika əsas rol oynayır. Eşzamanlı amillər yalnız heterozigot xəstələrdə, yəni qüsurlu genin yalnız bir valideyndən ötürüldüyü xəstələrdə vacibdir. Homoziqot xəstələrdə patologiyanın özü ağırdır və yüksək ixtisaslı müalicə tələb edir.
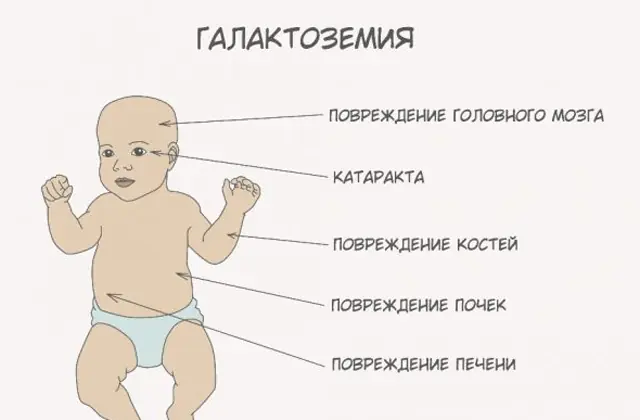
Qalaktozemiyanın inkişafının simptomları
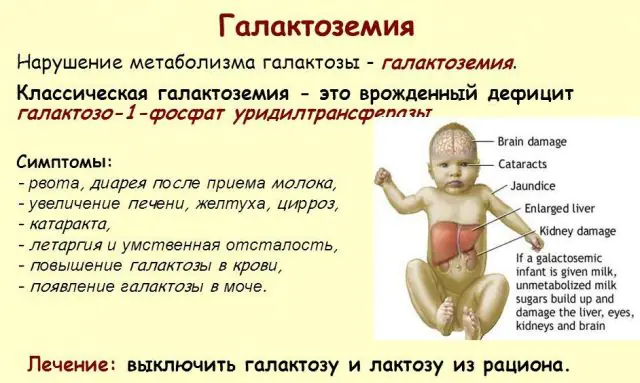
Qalaktozemiya əlamətlərinin təzahürlərinin şiddətinə görə xəstəliyin üç dərəcəsi fərqlənir:
- işıq- adətən süd dözümsüzlüyü səbəbindən təsadüfən aşkar edilir;
- orta- ilk əlamətlər yalnız süd içdikdən sonra görünür;
- ağır— patologiyanın simptomları bədəndəki digər nasazlıqlara (qarın boşluğunda mayenin yığılması, sepsis) əlavə olunur.
Klassik GALT patologiyası özünü ən ağır formada göstərir. Xəstəliyin ilk əlamətləri uşaq doğulduqdan və ilk qidalanmadan sonra görünəcək, çünki yeni doğulmuş körpənin əsas pəhrizini süd təşkil edir. Qidalandıqdan sonra o, daimi letarji və əzələ hipotenziyası fonunda qusma, tez-tez bağırsaq hərəkətləri və artan yuxululuq yaşayır. Müntəzəm olaraq ana südü ilə qidalanmaya baxmayaraq, çəki artımı yoxdur. Həzm pozğunluqları disfunksiyanın ilk əlamətlərindən biridir.
Orqanizm sərxoş olduqdan sonra qaraciyərin zədələnməsinin əlamətləri özünü büruzə verir - sarılıq, qaraciyərin böyüməsi. Bir neçə həftə ərzində uşaqda katarakt inkişaf edir və bir neçə aydan sonra sinir toxumalarının intoksikasiyası səbəbindən psixomotor funksiyaların pozulması qeyd olunur. Böyrək borucuqlarının fəaliyyəti pozulur, bunun nəticəsində sidikdə şəkərin azalması görünür və qan laxtalanmasının azalması qeyd olunur.
Lazımi terapiya olmadan, körpənin bədəni sərxoş olduqda, simptomlar pisləşir və normal inkişaf və hətta insan həyatı üçün risk artır. Uşaqların 20-30% -i müalicədən imtina edildikdə və ya xəstəlik gec diaqnoz qoyulduqda, leykositotik aktivliyə görə inkişaf edən sepsisdən ölür. Sağ qalanlar böyrək çatışmazlığından və psixomotor inkişaf problemlərindən əziyyət çəkirlər.
Bir çox cəhətdən, patologiyanın təzahür dərəcəsi irsi səbəbdən asılıdır. Qalaktozemiya GALT çatışmazlığının meydana gəlməsi ilə özünü göstərir. Beləliklə, Duhart əlaməti əvvəlcə özünü yalnız uzun müddətli sarılıq (doğuşdan sonra 2 aya qədər), katarakta və qaraciyər disfunksiyası kimi bir az sonra aşkar edə bilər. Ancaq GALT fermentinin aktivliyi ən azı 50% olarsa, klinik simptomlar görünməyə bilər.
GALA fermentinin çatışmazlığı o qədər də aydın deyil. Xəstəliyin əsas əlaməti mütərəqqi kataraktadır və zaman keçdikcə sidikdə monosaxaridlər də görünə bilər. Eyni zamanda, körpənin kilo itkisi əhəmiyyətsizdir və psixomotor bacarıqlar normal diapazonda inkişaf edir.
GALE çatışmazlığı başlanğıcda olduqca nadirdir, lakin simptomlar hələ də iki formaya bölünür: ferment çatışmazlığı qan hüceyrələrinə təsir etdikdə xoşxassəli və çatışmazlıq bədənin bütün toxumalarına təsir etdikdə ümumiləşdirilmişdir. Birinci halda, yalnız qalaktozemiya üçün bir analiz xəstəliyi erkən mərhələlərdə aşkar etməyə kömək edəcək, yalnız zamanla psixomotor bacarıqların və nitqin inkişafının pozulması qeyd olunacaq. İkinci forma simptomlarına görə GALT çatışmazlığına bənzəyir, lakin bu halda dalağın böyüməsi də var. GALE xəstəliyinin ağır formasının müalicəsi ilə belə, zaman keçdikcə uşaqda gecikmiş psixomotor inkişaf, karlıq və görmə problemləri yaranır.
Qalaktozemiya diaqnozu
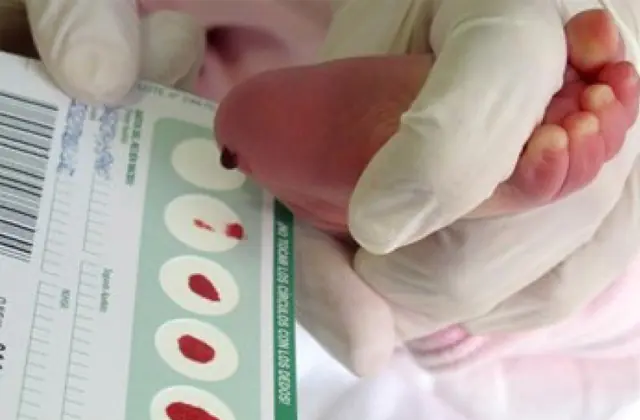
Bir sıra ölkələrdə tətbiq edilən neonatal skrininq ilkin mərhələdə 50-yə qədər anadangəlmə xəstəliyi, o cümlədən maddələr mübadiləsində fəal iştirak edən fermentlərin kifayət qədər istehsal olunmamasını müəyyən etməyə imkan verir. Hər hansı bir səbəbdən skrininq aparılmırsa, xəstəlik ümumi xüsusiyyətlərə əsasən müəyyən edilir.
Beləliklə, əgər qalaktozemiyadan şübhələnirsinizsə, həkim tövsiyələri yalnız ətraflı diaqnozdan sonra verilir. Kiçik bir xəstə aşağıdakılardan keçməlidir:
- ümumi müayinə - sarılıq əlamətlərinin olub-olmaması;
- ümumi əlamətlər qiymətləndirilir - bağırsaq hərəkətlərinin miqdarı və keyfiyyəti, əzələ hipotonikliyinin olması və ya olmaması, psixomotor bacarıqlarla bağlı problemlər;
- laboratoriya testləri - biokimya üçün qan testi, ümumi qan testi, şəkər və protein üçün sidik testi;
- qaraciyər və böyrəklərin vəziyyətinin əlavə müayinəsi.
GALT mutasiyalarını aşkar etmək üçün DNT diaqnostikası məcburidir. Uşaqda xəstəliyin simptomları varsa və test nəticələrinə görə GALT fermentlərinin aktivliyi normaldırsa, GALE fermenti üçün DNT diaqnostikası aparılır.
Diaqnozu fərqləndirmək üçün həkimlər bir sıra əlavə testlər təyin edə bilərlər. Diabetes mellitus və yüksək şəkər səviyyəsi ilə əlaqəli digər patologiyaların, həmçinin qaraciyərin genişlənməsinə səbəb olan xəstəliklərin mövcudluğunu təsdiqləmək və ya təkzib etmək üçün eksklüziv diaqnoz lazımdır.
Qalaktozemiyanı necə müalicə etmək olar?

Neonatal skrininq və digər müayinələr qalaktozemiyanın mövcudluğunu təsdiq etdikdən sonra müalicə təyin edilir. Anlamaq lazımdır ki, xəstəlik genetik meyldən qaynaqlandığı üçün tibbi inkişafın bu mərhələsində tam müalicə mümkün deyil. Terapiya patologiyanın klinik təzahürlərini azaltmağa, xəstəliyin gedişatını və ağırlaşmaların inkişafını minimuma endirməyə yönəldilmişdir.
Əsas müalicə üsulu qalaktozemiya üçün düzgün qidalanmadır. Bütün süd tərkibli məhsullar, hətta çörək, şirniyyat, kolbasa və digərləri xəstənin pəhrizindən tamamilə xaric edilir. Oliqosakaridləri olan bitki və heyvan mənşəli məhsullar da çıxarılır: paxlalılar, soya, ispanaq, şokolad, qoz-fındıq, qaraciyər, beyinlər, yumurtalar və bu məhsulların törəmələri. Ciddi pəhriz terapiyası bədənin intoksikasiyasının qarşısını almağın yeganə yoludur.
Yenidoğulmuşlar üçün qalaktozemiya üçün qidalanma da ixtisaslaşmışdır. Ana südü tədricən pəhrizdən tamamilə çıxarılır və soya proteini izolatına əsaslanan laktozasız formulalar və formulalar ilə əvəz olunur. Bu vəziyyətdə qida maddələrinin həcmi sağlam uşaq üçün standartlara uyğun olmalıdır. Körpənin soya zülalının izolatına allergiya əlamətləri varsa, kazein hidrolizatlarına əsaslanan qarışıqlar seçilir. Qida qarışığının ilk qəbulu uşağın gündəlik qida ehtiyacının 1/10 hissəsi olmalıdır. 5-7 gün ərzində ana südü pəhrizdən tamamilə xaric edilir.
Patoloji olan uşaqlar üçün ilk əlavə qidalar 4 aydan etibarən təyin edilir. Pəhriz meyvə şirələri (alma, armud və başqaları) ilə genişləndirilir. Yarım aydan sonra meyvə püresi təqdim olunur. 1 ilə qədər gündə istehlak edilən şirənin və pürenin həcmi 30-50 ml-ə qədər artır. Suda ilk tərəvəz püresi (paxlalılar istisna olmaqla) 5 ayda təqdim olunur. 5,5 aydan etibarən uşağa südsüz ticarət dənli bitkilər (qarabaşaq, düyü, qarğıdalı) verilə bilər. Körpə 6 aylıq yaşdan etibarən ət məhsullarını sınaya bilər, süd və ya süd tərkibli məhsullar olmayan xüsusi konservləşdirilmiş qidalara üstünlük verilməlidir.
Körpələr üçün xüsusi qida məhsulları seçərkən etiketləri diqqətlə oxumalısınız. 100 q-da 5 mq-dan çox olmayan qalaktoza olan məhsullar körpə üçün təhlükəsiz sayılır.20 mq-dan çox qalaktoza varsa, məhsulu uşağa vermək qəti qadağandır.
Qalaktozemiya üçün müalicənin adekvatlığı rübdə bir dəfə nəzarət müayinəsi ilə yoxlanılır. Patologiyanın müşayiət olunan simptomlarının müalicəsinin effektivliyi də sınaqdan keçirilir.
Vacibdir! Belə xəstələr üçün dərman seçiminə xüsusi diqqət yetirilir. Bəzi dərmanlar tərkibində laktoza ehtiva edir, digərləri isə qaraciyərdən metabolik məhsulların çıxarılmasını maneə törədə bilər, bu, ümumiyyətlə anadangəlmə anomaliya əlamətlərini ağırlaşdırır və səlahiyyətli müalicə çərçivəsində qəbuledilməzdir.Qalaktozemiyanın qarşısının alınması

Xəstəliyə qarşı xüsusi profilaktik tədbirlər hələ hazırlanmamışdır. Valideynlərin patologiyaya meylli olduğu aşkar edilərsə, bir genetikçi ilə əlavə məsləhətləşmələrlə konsepsiyanı planlaşdırmaq tövsiyə olunur. Hamiləlik dövründə genlərdə GALT və GALE mutasiyalarını aşkar etmək üçün bir sıra müayinələrdən də keçməlisiniz (10-12 həftədə xorion villus biopsiyası və 15-18 həftədə amniotik maye testi).
Patologiyanın kifayət qədər effektiv simptomatik müalicəsinə baxmayaraq, uşağın inkişafının uzunmüddətli perspektivlərini proqnozlaşdırmaq çətindir. Beləliklə, xəstənin fiziki inkişaf göstəriciləri həmyaşıdlarından daha aşağı olacaq, nitq və koordinasiya pozğunluqları inkişaf edə bilər, sümük kövrəkliyi artır, qızlarda isə yumurtalıq disfunksiyası.
5 yaşından etibarən uşağa xəstəliyin inkişafının qarşısını almaq üçün vitaminlər və ATP tərkibli dərmanlar qəbul etmək tövsiyə oluna bilər. 12 yaşından etibarən qızlara yumurtalıq disfunksiyasını kompensasiya etmək üçün hormonal terapiya təyin edilir.
Qalaktozemiya genetik cəhətdən müəyyən edilmiş olduqca nadir bir xəstəlikdir. Patologiyanın inkişaf mexanizmi hələ hərtərəfli öyrənilməmişdir, lakin problemin ciddiliyi bir sıra effektiv diaqnostik üsulların inkişafına səbəb olmuşdur. Bir sıra ölkələrdə məcburi olan neonatal skrininq xəstəliyi vaxtında aşkar etməyə və tədbir görməyə imkan verir. Genetik olaraq təyin olunan xəstəliklərin müalicəsi bu günə qədər tam olaraq inkişaf etdirilməsə də, terapiya xəstəliyin ağır simptomlarından və onun irəliləməsindən xilas olmağa kömək edir. Bu vəziyyətdə valideynlərin vəzifəsi patologiyanı mümkün qədər erkən diaqnoz etmək və həkimin göstərişlərinə əməl etmək, bununla da uşağa anadangəlmə anomaliya ilə mübarizə aparmaqdır.
Qalaktozemiya nədir - videoya baxın:
