
Was ist Galaktosämie und warum tritt sie bei einem Kind auf? Symptome, Diagnose der Krankheit. Behandlung von Galaktosämie bei Neugeborenen und verfügbare vorbeugende Maßnahmen.
Der Inhalt des Artikels:- Was ist Galaktosämie?
- Entwicklungssymptome
- Diagnose
- Wie behandelt man Galaktosämie?
- Verhütung
Galaktosämie ist eine seltene, aber immer noch häufige erbliche Stoffwechselstörung. Enzymopathie ist mit einer Störung des Kohlenhydratstoffwechsels verbunden, wodurch der Körper Galaktose und seine Derivate ansammelt. Übermäßige Mengen an Galaktose führen zur Entwicklung eines Krankheitsbildes. Bei einer späten Diagnose einer Galaktosämie verstärken sich die Symptome und die Prognose für eine Normalisierung des Zustands des Patienten ist düster. Paare mit einer Veranlagung für die Krankheit müssen über die Arten der Galaktosämie, Diagnose-, Behandlungs- und Präventionsmethoden Bescheid wissen und auch über Familienplanung nachdenken. Durch rechtzeitige Maßnahmen wird das Risiko von Komplikationen beim Neugeborenen deutlich reduziert.
Was ist Galaktosämie?
Bevor über Empfehlungen bei Galaktosämie gesprochen wird, ist es notwendig, die Rolle von Galaktose im Kohlenhydratstoffwechsel zu verstehen. Dieser Einfachzucker gelangt als Teil der Laktose in den menschlichen Körper, einem Disaccharid, dessen zweiter Bestandteil Glukose ist. Normalerweise wird das Monosaccharid als „Energie“-Ressource für Zellen und komplexe Verbindungen sowie als „Baumaterial“ für Nervengewebe, Membranen und Nervenenden verwendet.
Bei einem gesunden Menschen wird Laktose im Verdauungstrakt in seine Bestandteile zerlegt und nach und nach verarbeitet. So wird in der ersten Verarbeitungsstufe Galactose-1-phosphat unter Einwirkung eines speziellen Enzyms Galactokinase und ATP gewonnen. Im nächsten Schritt werden durch den Austausch unter Einwirkung von Enzymen Glucose-1-phosphat und Uridinphosphat-Galactose gewonnen. Monosaccharide werden bei der Verdauung viel leichter vom Darm aufgenommen, was für Neugeborene sehr wichtig ist, da Milch die Grundlage ihrer Ernährung ist.
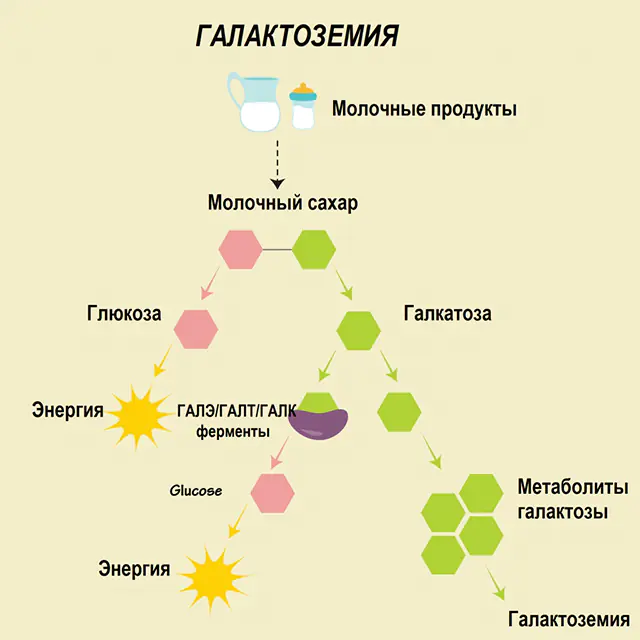
An den Verarbeitungsprozessen sind drei Enzyme beteiligt – GALA (Galactokinase), GALT (Galactose-1-phosphat-Uridyltransferase), GALE (Epimerase). Eine mangelhafte Produktion mindestens eines dieser Enzyme im ersten oder zweiten Stadium des Kohlenhydratstoffwechsels führt zur Ansammlung von Metaboliten, einer Vergiftung des Körpers und der anschließenden Manifestation von Galaktosämie-Symptomen.
Abhängig vom defekten Enzym werden die entsprechenden Formen der Galaktosämie unterschieden:
- GALT-Verstoß- Der erste Typ tritt in einem Fall pro 40.000 Neugeborenen auf; die Analyse auf Galaktosämie zeigt eine verminderte Enzymaktivität;
- GALC-Pathologie- Der zweite Typ tritt bei einem von 500.000 Kindern auf, während die Aktivität des Grundenzyms in Erythrozyten im normalen Bereich liegt, die Krankheitssymptome jedoch ausgeprägt sind;
- STURM— Der dritte Typ wurde am wenigsten registriert (ein Fall pro Million Menschen) und zeichnet sich durch milde Symptome aus.
Die häufigste Krankheitsart (GALT) wird je nach Genetik der Galaktosämie in folgende Formen eingeteilt:
- Das Versagen ist charakteristisch für das Stadium der Umwandlung von Asparaginsäureamid in Aspartat – die Pathologie wird auch Duarte-Zeichen (Duarte) genannt. Es ist bemerkenswert, dass sich eine Person mit einer solchen Anomalie absolut gesund fühlt, obwohl die Aktivität des GALT-Enzyms um 25–50 % reduziert ist.
- Ein Versagen des Glutamin-Arginin-Übergangs wird am häufigsten bei Kaukasiern beobachtet; die Enzymaktivität beträgt nur 10 % der Norm, was zu einem schweren Krankheitsverlauf führt.
- Eine Störung des Arginin-Tryptophan-Übergangs tritt auf, wenn das Enzym völlig inaktiv ist, eine äußerst schwere Form der Krankheit.
- Eine Störung des Lysin-Asparagin-Umwandlungsprozesses kommt häufig vor.
- Ein Versagen der Serin-Leucin-Fermentation wurde nur bei Vertretern der Negerrasse festgestellt und ist mit einer unzureichenden Enzymaktivität in der Leber verbunden (auf einem Niveau von nicht mehr als 10 % der allgemein anerkannten Norm).
Das GALT-Enzym ist im Körper in übermäßigen Mengen vorhanden, sodass sich eine leichte Abnahme seiner Aktivität nicht immer in klinischen Symptomen äußert. Erst wenn die GALT-Aktivität um 50 Prozent oder mehr sinkt, treten erste Krankheitszeichen auf.
Eine Vergiftung des Körpers mit Galaktose und Metaboliten ist auf lange Sicht gefährlich für die Entwicklung von Komplikationen: Beeinträchtigung der Motorik, Sprache, Fortpflanzungsfunktionen, insbesondere bei Mädchen (obwohl die Häufigkeit von Erkrankungen bei Jungen und Mädchen gleich ist), Wachstumsverzögerung und andere Entwicklungsstörungen.
Beachten Sie! Die Pathogenese der Krankheit ist noch nicht vollständig untersucht. Es wurde jedoch bereits festgestellt, dass die Anreicherung von Galaktose im Körper nicht nur wegen seiner toxischen Dosierung, sondern auch wegen seiner hemmenden Wirkung auf andere Enzyme gefährlich ist. Ohne angemessene Behandlung und Kontrolle kann der Patient ein hypoglykämisches Syndrom entwickeln.Wie bereits erwähnt, handelt es sich bei der Pathologie um eine angeborene Anomalie. Die Art der Vererbung der Galaktosämie ist autosomal-rezessiv, das heißt, das Kind erhält von den Eltern defekte Gene. Das mutierte Gen kann in verschiedenen Allelen (Erkrankungsformen) gefunden werden – defekt und normal, wenn der Defekt nur von einem Elternteil übertragen wurde. In diesem Fall manifestiert sich die Krankheit in geringerem Maße. Wenn jedoch defekte Allele entsprechend der Vererbungsart der Galaktosämie sowohl vom Vater als auch von der Mutter vererbt wurden, treten die ersten Krankheitszeichen bereits wenige Tage nach der Geburt des Kindes auf. Es ist bemerkenswert, dass, wenn bei einem Kind in einer Familie eine Krankheit diagnostiziert wird, die Wahrscheinlichkeit, dass die Pathologie beim nächsten Kind derselben Eltern auftritt, um 25 % steigt.
Eine unzureichende Produktion von Enzymen führt zur Anreicherung von Galaktose im Körper, wodurch Anzeichen einer Galaktosämie auftreten und die Funktionen des Ausscheidungssystems gestört werden. Ohne Behandlung stirbt eine Person innerhalb weniger Lebensmonate an Leberversagen oder Infektionen, die den Körper beeinträchtigen.
Es ist wichtig, dass die Hauptursachen der Galaktosämie (erbliche Komponente) bereits vor der Geburt des Kindes festgestellt werden können, Ärzte schließen jedoch den Einfluss anderer Indikatoren auf die Entwicklung der Pathologie nicht aus:
- andere Genmutationen;
- betroffenes Gewebe der Leber und des Zentralnervensystems;
- Ansammlung von Flüssigkeit in der Augenlinse, verschwommenes Sehen.
Und doch spielt die Genetik eine Schlüsselrolle, wenn bei einem Patienten Galaktosämie diagnostiziert wird. Begleitfaktoren spielen nur bei heterozygoten Patienten eine Rolle, also solchen, bei denen das defekte Gen nur von einem Elternteil vererbt wurde. Bei homozygoten Patienten ist die Pathologie selbst schwerwiegend und erfordert eine hochspezialisierte Behandlung.
Symptome der Entwicklung einer Galaktosämie
Je nach Schwere der Manifestationen der Anzeichen einer Galaktosämie werden drei Krankheitsgrade unterschieden:
- Licht- meist zufällig aufgrund einer Milchunverträglichkeit entdeckt;
- Durchschnitt- Die ersten Anzeichen treten erst nach dem Trinken von Milch auf;
- schwer— Die Symptome der Pathologie überlagern sich mit anderen Fehlfunktionen im Körper (Flüssigkeitsansammlung in der Bauchhöhle, Sepsis).
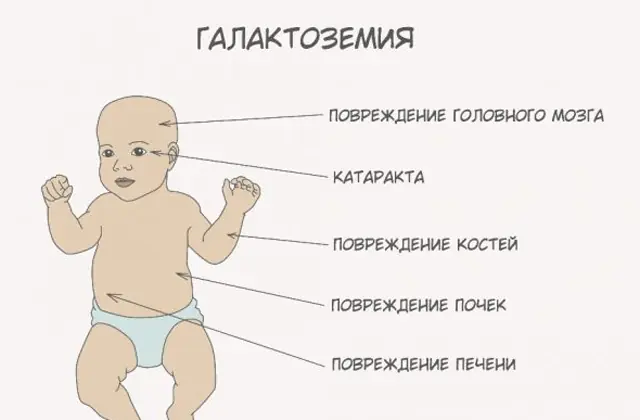
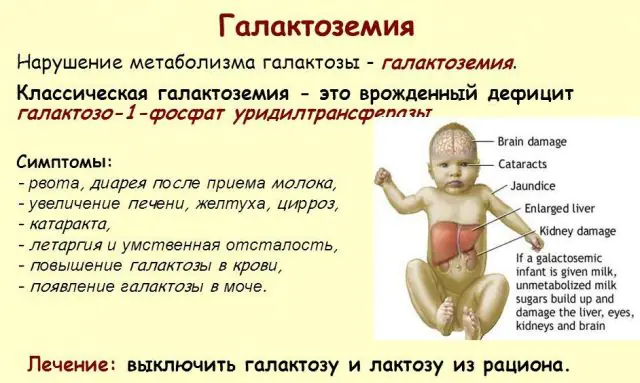
Die klassische GALT-Pathologie manifestiert sich in ihrer schwersten Form. Die ersten Anzeichen der Krankheit werden nach der Geburt des Kindes und der ersten Fütterung sichtbar, da Milch die Hauptnahrung des Neugeborenen darstellt. Nach dem Füttern verspürt er Erbrechen, häufigen Stuhlgang und erhöhte Schläfrigkeit vor dem Hintergrund ständiger Lethargie und Muskelhypotonie. Trotz regelmäßigem Stillen kommt es zu keiner Gewichtszunahme. Verdauungsstörungen sind eines der ersten Anzeichen einer Funktionsstörung.
Wenn der Körper berauscht wird, treten deutliche Anzeichen einer Leberschädigung auf – Gelbsucht, Lebervergrößerung. Innerhalb weniger Wochen entwickelt das Kind einen Katarakt und nach einigen Monaten werden aufgrund einer Vergiftung des Nervengewebes Störungen der psychomotorischen Funktionen festgestellt. Die Aktivität der Nierentubuli ist gestört, wodurch reduzierender Zucker im Urin auftritt und eine verminderte Blutgerinnung festgestellt wird.
Ohne die notwendige Therapie verschlimmern sich die Symptome, da der Körper des Babys vergiftet wird und das Risiko für die normale Entwicklung und sogar das menschliche Leben steigt. 20-30 % der Kinder sterben an einer Sepsis, die sich aufgrund der leukozytotischen Aktivität entwickelt, wenn die Behandlung verweigert oder die Krankheit zu spät diagnostiziert wird. Die Überlebenden leiden unter Nierenversagen und psychomotorischen Entwicklungsstörungen.
Der Grad der Manifestation der Pathologie hängt in vielerlei Hinsicht von der erblichen Ursache ab. Galaktosämie äußert sich in einem GALT-Mangel. So kann sich das Duhart-Zeichen zunächst nur als anhaltende Gelbsucht (bis zu 2 Monate nach der Geburt) äußern, wenig später werden Katarakte und Leberfunktionsstörungen diagnostiziert. Wenn die Aktivität des GALT-Enzyms jedoch mindestens 50 % beträgt, treten möglicherweise keine klinischen Symptome auf.
Der Mangel an GALA-Enzym ist nicht so ausgeprägt. Das Hauptsymptom der Krankheit sind fortschreitende Katarakte, im Laufe der Zeit können auch Monosaccharide im Urin auftreten. Gleichzeitig ist der Gewichtsverlust des Babys unbedeutend und die psychomotorischen Fähigkeiten entwickeln sich im normalen Bereich.
GALE-Mangel ist anfangs äußerst selten, die Symptome werden jedoch noch in zwei Formen unterteilt: gutartig, wenn der Mangel des Enzyms die Blutzellen betrifft, und generalisiert, wenn der Mangel alle Gewebe des Körpers betrifft. Im ersten Fall hilft nur eine Analyse auf Galaktosämie, die Krankheit im Frühstadium zu erkennen; erst mit der Zeit werden Störungen der psychomotorischen Fähigkeiten und der Sprachentwicklung festgestellt. Die zweite Form ähnelt in ihren Symptomen dem GALT-Mangel, allerdings kommt es auch hier zu einer Vergrößerung der Milz. Selbst bei der Behandlung einer schweren Form der GALE-Krankheit kommt es im Laufe der Zeit zu einer verzögerten psychomotorischen Entwicklung, Taubheit und Sehstörungen.
Diagnose einer Galaktosämie
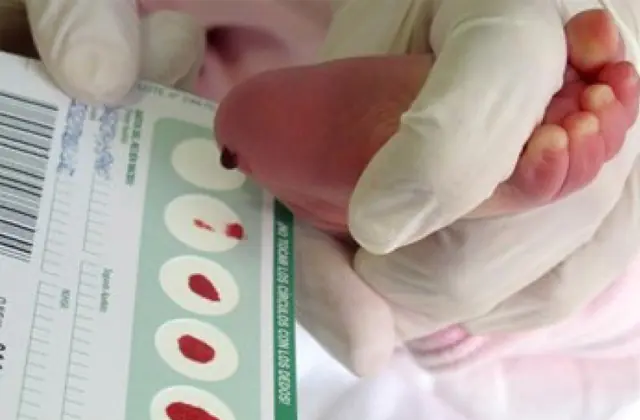
Das in mehreren Ländern eingeführte Neugeborenen-Screening ermöglicht die Erkennung von bis zu 50 angeborenen Krankheiten im Frühstadium, darunter eine unzureichende Produktion von Enzymen, die aktiv am Stoffwechsel beteiligt sind. Wenn aus irgendeinem Grund kein Screening durchgeführt wird, wird die Krankheit anhand aggregierter Merkmale identifiziert.
Bei Verdacht auf Galaktosämie werden ärztliche Empfehlungen also erst nach einer ausführlichen Diagnose abgegeben. Ein kleiner Patient muss sich unterziehen:
- allgemeine Untersuchung – ob Anzeichen einer Gelbsucht vorliegen;
- allgemeine Anzeichen werden beurteilt – Quantität und Qualität des Stuhlgangs, Vorhandensein oder Fehlen einer Muskelhypotonie, Probleme mit psychomotorischen Fähigkeiten;
- Labortests – Bluttest für Biochemie, allgemeiner Bluttest, Urintest für Zucker und Protein;
- zusätzliche Untersuchung des Zustands von Leber und Nieren.
Zum Nachweis von GALT-Mutationen ist eine DNA-Diagnostik zwingend erforderlich. Wenn das Kind Krankheitssymptome aufweist und die Aktivität der GALT-Enzyme laut Testergebnissen normal ist, wird eine DNA-Diagnostik für das GALE-Enzym durchgeführt.
Ärzte können eine Reihe zusätzlicher Tests verschreiben, um die Diagnose zu differenzieren. Eine ausschließliche Diagnose ist erforderlich, um das Vorliegen von Diabetes mellitus und anderen Pathologien, die mit einem hohen Zuckerspiegel einhergehen, sowie von Krankheiten, die eine Lebervergrößerung verursachen, zu bestätigen oder zu widerlegen.
Wie behandelt man Galaktosämie?

Sobald das Neugeborenen-Screening und andere Untersuchungen das Vorliegen einer Galaktosämie bestätigt haben, wird eine Behandlung verordnet. Man muss verstehen, dass eine vollständige Heilung in diesem Stadium der medizinischen Entwicklung unmöglich ist, da die Krankheit durch eine genetische Veranlagung verursacht wird. Die Therapie zielt darauf ab, die klinischen Manifestationen der Pathologie zu reduzieren, das Fortschreiten der Krankheit und die Entwicklung von Komplikationen zu minimieren.
Die wichtigste Behandlungsmethode ist die richtige Ernährung bei Galaktosämie. Alle milchhaltigen Produkte, auch Brot, Süßigkeiten, Wurstwaren und andere, sind vom Speiseplan des Patienten komplett ausgeschlossen. Auch Produkte pflanzlichen und tierischen Ursprungs, die Oligosaccharide enthalten, werden entfernt: Hülsenfrüchte, Soja, Spinat, Schokolade, Nüsse, Leber, Gehirne, Eier und Derivate dieser Produkte. Nur durch eine strenge Ernährungstherapie kann eine Vergiftung des Körpers verhindert werden.
Auf Neugeborene ist auch die Ernährung bei Galaktosämie spezialisiert. Muttermilch wird nach und nach vollständig aus der Ernährung gestrichen und durch laktosefreie Säuglingsnahrung und Zubereitungen auf Basis von Sojaproteinisolat ersetzt. Die Menge an Nährstoffen sollte in diesem Fall den Standards für ein gesundes Kind entsprechen. Wenn ein Baby Symptome einer Allergie gegen Sojaproteinisolat zeigt, werden Mischungen auf Basis von Kaseinhydrolysaten ausgewählt. Die erste Nahrungsmischung sollte 1/10 des täglichen Nährstoffbedarfs des Kindes betragen. Innerhalb von 5-7 Tagen wird die Muttermilch vollständig aus der Ernährung gestrichen.
Die ersten Ergänzungsnahrungsmittel für Kinder mit Pathologie werden ab dem 4. Monat verschrieben. Die Ernährung wird durch Fruchtsäfte (Apfel, Birne und andere) erweitert. Nach einem halben Monat werden Fruchtpürees eingeführt. Bis zu einem Jahr erhöht sich die täglich konsumierte Saft- und Püreemenge auf 30-50 ml. Das erste Gemüsepüree in Wasser (außer Hülsenfrüchte) wird mit 5 Monaten eingeführt. Ab 5,5 Monaten kann ein Kind milchfreies kommerzielles Getreide (Buchweizen, Reis, Mais) erhalten. Ab einem Alter von 6 Monaten kann das Baby Fleischprodukte probieren, wobei spezielle Konserven, die keine Milch oder milchhaltige Produkte enthalten, bevorzugt werden sollten.
Bei der Auswahl spezieller Lebensmittel für Kleinkinder müssen Sie die Etiketten sorgfältig lesen. Produkte, die nicht mehr als 5 mg Galaktose pro 100 g enthalten, gelten als sicher für ein Baby. Bei mehr als 20 mg Galaktose ist die Verabreichung des Produkts an ein Kind strengstens verboten.
Die Angemessenheit der Behandlung der Galaktosämie wird vierteljährlich durch eine Kontrolluntersuchung überprüft. Die Wirksamkeit der Behandlung begleitender pathologischer Symptome wird ebenfalls getestet.
Wichtig! Besonderes Augenmerk wird auf die Auswahl der Medikamente für solche Patienten gelegt. Einige Medikamente enthalten in ihrer Zusammensetzung Laktose, andere können den Abtransport von Stoffwechselprodukten aus der Leber hemmen, was im Allgemeinen die Symptome der angeborenen Anomalie verschlimmert und im Rahmen einer kompetenten Behandlung nicht akzeptabel ist.Vorbeugung von Galaktosämie

Spezielle Präventionsmaßnahmen für die Krankheit wurden noch nicht entwickelt. Wenn bei den Eltern eine Veranlagung für die Pathologie festgestellt wurde, wird empfohlen, die Empfängnis mit zusätzlichen Konsultationen mit einem Genetiker zu planen. Während der Schwangerschaft sollten Sie sich außerdem einer Reihe von Untersuchungen (Chorionzottenbiopsie in der 10.–12. Woche und Fruchtwassertest in der 15.–18. Woche) unterziehen, um GALT- und GALE-Mutationen in Genen festzustellen.
Trotz der entwickelten recht wirksamen symptomatischen Behandlung der Pathologie sind die langfristigen Aussichten für die Entwicklung des Kindes schwer vorherzusagen. Daher sind die körperlichen Entwicklungsindikatoren der Patientin niedriger als die ihrer Altersgenossen, es kommt wahrscheinlich zu Sprach- und Koordinationsstörungen, erhöhter Knochenbrüchigkeit und bei Mädchen zu einer Funktionsstörung der Eierstöcke.
Ab dem 5. Lebensjahr kann einem Kind die Einnahme von Vitaminen und ATP-haltigen Medikamenten empfohlen werden, um das Fortschreiten der Krankheit zu verhindern. Ab dem 12. Lebensjahr wird Mädchen eine Hormontherapie verschrieben, um eine Funktionsstörung der Eierstöcke auszugleichen.
Galaktosämie ist eine relativ seltene genetisch bedingte Krankheit. Der Entstehungsmechanismus der Pathologie wurde noch nicht gründlich untersucht, aber die Schwere des Problems hat zur Entwicklung einer Reihe wirksamer Diagnosetechniken geführt. Das in vielen Ländern obligatorische Neugeborenen-Screening ermöglicht es, die Krankheit rechtzeitig zu erkennen und Maßnahmen zu ergreifen. Und obwohl die Behandlung genetisch bedingter Krankheiten bis heute noch nicht ausgereift ist, hilft die Therapie, schwere Krankheitssymptome und deren Fortschreiten zu beseitigen. Die Aufgabe der Eltern besteht in diesem Fall darin, die Pathologie so früh wie möglich zu diagnostizieren und den Anweisungen des Arztes zu folgen und so dem Kind bei der Bewältigung der angeborenen Anomalie zu helfen.
Was ist Galaktosämie? Sehen Sie sich das Video an:
