
Apa itu galaktosemia dan mengapa hal itu terjadi pada anak-anak? Gejala, diagnosis penyakit. Pengobatan galaktosemia pada bayi baru lahir dan tindakan pencegahan yang tersedia.
Isi artikel:- Apa itu galaktosemia
- Gejala perkembangan
- Diagnostik
- Cara mengobati galaktosemia
- Pencegahan
Galaktosemia adalah kelainan herediter yang berhubungan dengan metabolisme yang jarang namun masih umum terjadi. Enzimopati dikaitkan dengan disfungsi metabolisme karbohidrat, akibatnya tubuh menumpuk galaktosa dan turunannya. Jumlah galaktosa yang berlebihan menyebabkan perkembangan gambaran klinis. Jika diagnosis galaktosemia terlambat, gejalanya menjadi jelas, dan prognosis untuk normalisasi kondisi pasien suram. Pasangan yang memiliki kecenderungan penyakit ini perlu mengetahui jenis-jenis galaktosemia, cara diagnosis, pengobatan dan pencegahan, serta memikirkan tentang keluarga berencana. Tindakan tepat waktu yang diambil secara signifikan mengurangi risiko komplikasi pada bayi baru lahir.
Apa itu galaktosemia?
Sebelum berbicara tentang rekomendasi untuk galaktosemia, perlu dipahami peran galaktosa dalam metabolisme karbohidrat. Gula sederhana ini masuk ke dalam tubuh manusia sebagai bagian dari laktosa, suatu disakarida yang komponen kedua adalah glukosa. Biasanya, monosakarida digunakan sebagai sumber “energi” untuk sel dan senyawa kompleks, bahan “pembangun” jaringan saraf, membran, dan ujung saraf.
Pada orang sehat, laktosa dipecah menjadi komponen-komponennya di saluran pencernaan dan diproses secara bertahap. Jadi, pada tahap pertama pemrosesan, galaktosa 1-fosfat diperoleh di bawah aksi enzim khusus galaktokinase dan ATP. Pada tahap selanjutnya, sebagai hasil pertukaran di bawah aksi enzim, glukosa 1-fosfat dan uridin fosfat galaktosa diperoleh. Monosakarida jauh lebih mudah diserap oleh usus selama pencernaan, yang sangat penting bagi bayi baru lahir, karena susu adalah makanan pokok mereka.
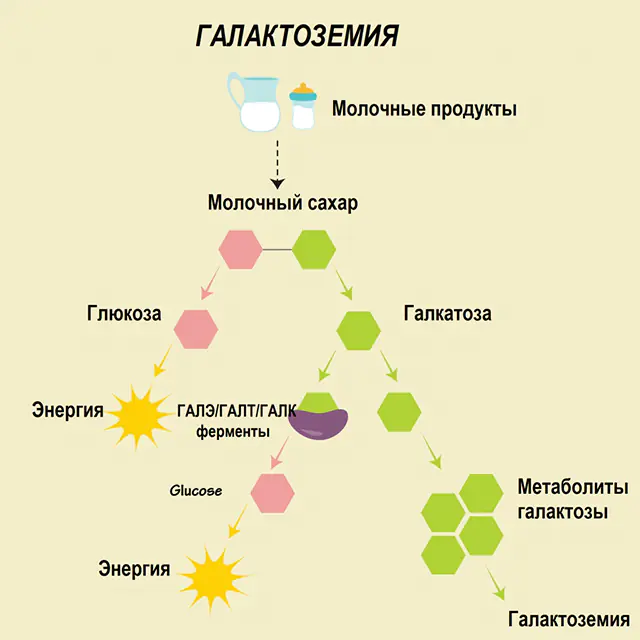
Tiga enzim terlibat dalam proses pemrosesan - GALA (galaktokinase), GALT (galaktosa 1-fosfat uridiil transferase), GALE (epimerase). Kekurangan produksi setidaknya satu dari enzim tersebut pada tahap pertama atau kedua metabolisme karbohidrat menyebabkan akumulasi metabolit, keracunan tubuh, dan manifestasi gejala galaktosemia selanjutnya.
Tergantung pada enzim yang rusak, jenis galaktosemia yang sesuai dibedakan:
- pelanggaran GALT- tipe pertama terjadi pada satu kasus per 40 ribu bayi baru lahir, analisis galaktosemia akan menunjukkan penurunan aktivitas enzim;
- Patologi GALC- tipe kedua terjadi pada satu dari 500 ribu anak, sedangkan aktivitas enzim dasar dalam eritrosit berada dalam kisaran normal, tetapi gejala penyakitnya jelas;
- BADAI— tipe ketiga tercatat paling sedikit (satu kasus per juta orang) dan ditandai dengan gejala ringan.
Jenis penyakit yang paling umum (GALT) dibagi menjadi beberapa bentuk, tergantung pada genetika galaktosemia:
- Kegagalan tersebut merupakan ciri dari tahap transformasi asam aspartat tengah menjadi aspartat - patologinya juga disebut tanda Duarte (Duarte). Patut dicatat bahwa seseorang dengan kelainan seperti itu akan merasa benar-benar sehat, meskipun aktivitas enzim GALT berkurang 25%-50%.
- Kegagalan transisi glutamin-arginin paling sering tercatat di antara orang Kaukasia, aktivitas enzim hanya 10% dari normal, yang memicu perjalanan penyakit yang parah.
- Gangguan transisi arginin-triptofan terjadi ketika enzim benar-benar tidak aktif, suatu bentuk penyakit yang sangat parah.
- Gangguan pada proses transformasi lisin-asparagin cukup sering terjadi.
- Kegagalan fermentasi serin-leusin hanya tercatat pada perwakilan ras Negroid dan dikaitkan dengan aktivitas enzim yang tidak mencukupi di hati (pada tingkat tidak lebih dari 10% dari norma yang berlaku umum).
Enzim GALT terdapat dalam tubuh dalam jumlah berlebihan, sehingga sedikit penurunan aktivitasnya tidak selalu bermanifestasi sebagai gejala klinis. Hanya ketika aktivitas GALT turun 50 persen atau lebih, tanda-tanda pertama penyakit akan muncul.
Keracunan tubuh dengan galaktosa dan metabolitnya berbahaya bagi perkembangan komplikasi dalam jangka panjang: gangguan fungsi motorik, bicara, fungsi reproduksi, terutama pada anak perempuan (walaupun frekuensi penyakit pada anak laki-laki dan perempuan sama), keterbelakangan pertumbuhan dan disfungsi perkembangan lainnya.
Catatan! Patogenesis penyakit ini belum sepenuhnya dipahami. Namun, telah diketahui bahwa akumulasi galaktosa dalam tubuh berbahaya bukan hanya karena dosisnya yang toksik, tetapi juga karena efek penghambatannya terhadap enzim lain. Tanpa pengobatan dan kontrol yang tepat, pasien dapat mengalami sindrom hipoglikemik.Seperti telah disebutkan, patologinya adalah kelainan bawaan. Jenis pewarisan galaktosemia adalah autosomal resesif, yaitu anak menerima gen yang cacat dari orang tuanya. Gen yang bermutasi dapat ditemukan di alel yang berbeda (bentuk kondisi) - cacat dan normal, bila cacat tersebut diturunkan hanya dari satu orang tua. Dalam hal ini, penyakit ini memanifestasikan dirinya pada tingkat yang lebih rendah. Namun jika alel yang cacat telah diturunkan baik dari ayah maupun ibu sesuai dengan jenis pewarisan galaktosemia, maka tanda-tanda awal penyakit akan muncul dalam beberapa hari setelah bayi lahir. Patut dicatat bahwa jika suatu penyakit didiagnosis pada satu anak dalam sebuah keluarga, kemungkinan terjadinya patologi pada anak berikutnya dari orang tua yang sama meningkat sebesar 25%.
Produksi enzim yang tidak mencukupi menyebabkan penumpukan galaktosa dalam tubuh, akibatnya muncul tanda-tanda galaktosemia dan terganggunya fungsi sistem ekskresi. Tanpa pengobatan dalam beberapa bulan kehidupan, seseorang meninggal karena gagal hati atau infeksi yang mempengaruhi tubuh.
Penting bahwa penyebab utama galaktosemia (komponen keturunan) dapat diketahui bahkan sebelum kelahiran anak, namun dokter tidak mengecualikan pengaruh indikator lain pada perkembangan patologi:
- mutasi gen lainnya;
- jaringan hati dan sistem saraf pusat yang terkena;
- penumpukan cairan di lensa mata, penglihatan kabur.
Namun, jika seorang pasien didiagnosis menderita galaktosemia, faktor genetik memainkan peran kuncinya. Faktor penyerta hanya penting pada pasien heterozigot, yaitu pasien yang gen cacatnya diturunkan hanya dari satu orang tua. Pada pasien homozigot, patologinya sendiri parah dan memerlukan perawatan yang sangat khusus.
Gejala perkembangan galaktosemia
Menurut tingkat keparahan manifestasi tanda-tanda galaktosemia, tiga derajat penyakit dibedakan:
- lampu- biasanya terdeteksi secara tidak sengaja karena intoleransi susu;
- rata-rata- tanda-tanda pertama hanya muncul setelah minum susu;
- berat— gejala patologi ditumpangkan pada malfungsi lain dalam tubuh (akumulasi cairan di rongga perut, sepsis).
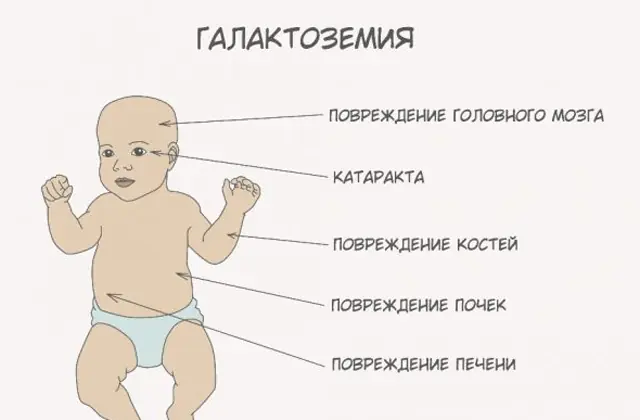
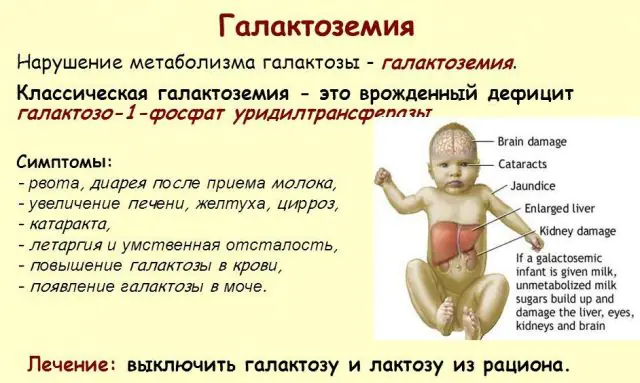
Patologi GALT klasik memanifestasikan dirinya dalam bentuk yang paling parah. Tanda-tanda pertama penyakit ini akan terlihat setelah bayi lahir dan menyusui pertamanya, karena susu merupakan makanan utama bayi baru lahir. Setelah makan, ia mengalami muntah-muntah, sering buang air besar, dan rasa kantuk yang meningkat dengan latar belakang kelesuan terus-menerus dan hipotensi otot. Meskipun menyusui secara teratur, tidak ada penambahan berat badan. Gangguan pencernaan adalah salah satu tanda pertama disfungsi.
Saat tubuh menjadi mabuk, tanda-tanda kerusakan hati menjadi jelas - penyakit kuning, pembesaran hati. Dalam beberapa minggu, anak mengalami katarak, dan setelah beberapa bulan, karena keracunan jaringan saraf, terjadi gangguan fungsi psikomotorik. Aktivitas tubulus ginjal terganggu, akibatnya terjadi penurunan gula dalam urin, dan penurunan pembekuan darah.
Tanpa terapi yang diperlukan, gejalanya akan memburuk karena keracunan tubuh bayi, dan risiko terhadap perkembangan normal dan bahkan kehidupan manusia meningkat. 20-30% anak-anak, jika pengobatan ditolak atau penyakitnya terlambat didiagnosis, meninggal karena sepsis, yang berkembang karena aktivitas leukositosis. Mereka yang bertahan hidup menderita gagal ginjal dan masalah perkembangan psikomotorik.
Dalam banyak hal, tingkat manifestasi patologi bergantung pada penyebab keturunan. Galaktosemia memanifestasikan dirinya ketika terjadi defisiensi GALT. Jadi, tanda Duhart pada awalnya hanya dapat muncul sebagai penyakit kuning yang berkepanjangan (hingga 2 bulan setelah lahir), katarak, dan disfungsi hati yang didiagnosis beberapa saat kemudian. Namun jika aktivitas enzim GALT minimal 50%, gejala klinis mungkin tidak muncul.
Kekurangan enzim GALA tidak begitu terasa. Gejala utama penyakit ini adalah katarak progresif, dan monosakarida juga dapat muncul dalam urin seiring berjalannya waktu. Pada saat yang sama, penurunan berat badan bayi tidak signifikan, dan keterampilan psikomotorik berkembang dalam batas normal.
Defisiensi GALE pada awalnya sangat jarang terjadi, namun gejalanya masih terbagi menjadi dua bentuk: jinak, bila kekurangan enzim mempengaruhi sel darah, dan umum, bila defisiensi mempengaruhi seluruh jaringan tubuh. Dalam kasus pertama, hanya analisis galaktosemia yang akan membantu mendeteksi penyakit pada tahap awal, hanya seiring berjalannya waktu disfungsi keterampilan psikomotorik dan perkembangan bicara akan diketahui. Bentuk kedua gejalanya mirip dengan defisiensi GALT, tetapi dalam kasus ini juga terjadi pembesaran limpa. Bahkan dengan pengobatan penyakit GALE yang parah, lama kelamaan anak tersebut mengalami keterlambatan perkembangan psikomotorik, ketulian, dan masalah penglihatan.
Diagnosis galaktosemia
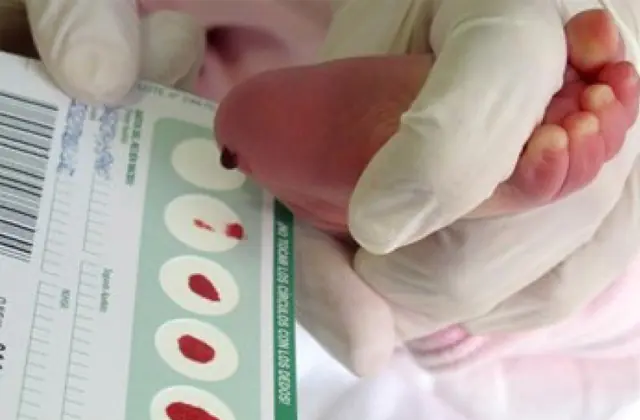
Skrining neonatal, yang diperkenalkan di sejumlah negara, memungkinkan untuk mengidentifikasi hingga 50 penyakit bawaan pada tahap awal, termasuk kurangnya produksi enzim yang secara aktif terlibat dalam metabolisme. Jika karena alasan tertentu skrining tidak dilakukan, penyakit tersebut diidentifikasi berdasarkan karakteristik agregat.
Jadi, jika dicurigai adanya galaktosemia, rekomendasi dokter dibuat hanya setelah diagnosis rinci. Seorang pasien kecil harus menjalani:
- pemeriksaan umum - apakah ada tanda-tanda penyakit kuning;
- tanda-tanda umum dinilai - kuantitas dan kualitas buang air besar, ada tidaknya hipotonisitas otot, masalah dengan keterampilan psikomotorik;
- tes laboratorium - tes darah untuk biokimia, tes darah umum, tes urin untuk gula dan protein;
- pemeriksaan tambahan terhadap kondisi hati dan ginjal.
Diagnostik DNA wajib untuk mendeteksi mutasi GALT. Jika anak menunjukkan gejala penyakit, dan aktivitas enzim GALT menurut hasil tes normal, maka dilakukan diagnosa DNA untuk enzim GALE.
Dokter mungkin meresepkan sejumlah tes tambahan untuk membedakan diagnosis. Diagnosis eksklusif diperlukan untuk memastikan atau menyangkal adanya diabetes mellitus dan patologi lain yang berhubungan dengan kadar gula tinggi, serta penyakit yang menyebabkan pembesaran hati.
Bagaimana cara mengobati galaktosemia?

Setelah skrining neonatal dan pemeriksaan lain memastikan adanya galaktosemia, pengobatan ditentukan. Perlu dipahami bahwa karena penyakit ini disebabkan oleh kecenderungan genetik, penyembuhan total pada tahap perkembangan medis ini tidak mungkin dilakukan. Terapi ditujukan untuk mengurangi manifestasi klinis patologi, meminimalkan perkembangan penyakit dan perkembangan komplikasi.
Metode pengobatan utama adalah nutrisi yang tepat untuk galaktosemia. Semua produk yang mengandung susu, bahkan roti, permen, sosis, dan lainnya, sama sekali tidak termasuk dalam makanan pasien. Produk nabati dan hewani yang mengandung oligosakarida juga dihilangkan: kacang-kacangan, kedelai, bayam, coklat, kacang-kacangan, hati, otak, telur, dan turunan dari produk tersebut. Terapi diet ketat adalah satu-satunya cara untuk mencegah keracunan pada tubuh.
Untuk bayi baru lahir, nutrisi untuk galaktosemia juga dikhususkan. ASI secara bertahap dihilangkan sepenuhnya dari makanan dan diganti dengan susu formula bebas laktosa dan formulasi berdasarkan isolat protein kedelai. Volume bahan gizi dalam hal ini harus memenuhi standar kesehatan anak. Jika bayi menunjukkan gejala alergi terhadap isolat protein kedelai, campuran berdasarkan hidrolisat kasein dipilih. Asupan pertama campuran makanan sebaiknya 1/10 dari kebutuhan gizi harian anak. Dalam waktu 5-7 hari, ASI dikeluarkan sepenuhnya dari makanan.
Makanan pendamping pertama untuk anak-anak dengan patologi diresepkan mulai 4 bulan. Dietnya diperluas dengan jus buah (apel, pir, dan lainnya). Setelah setengah bulan, pure buah diperkenalkan. Hingga 1 tahun, volume jus dan puree yang dikonsumsi per hari meningkat menjadi 30-50 ml. Haluskan sayuran pertama dalam air (kecuali kacang-kacangan) diperkenalkan pada usia 5 bulan. Mulai 5,5 bulan, seorang anak dapat diberikan sereal komersial bebas susu (gandum, nasi, jagung). Bayi dapat mencoba produk daging sejak usia 6 bulan, preferensi harus diberikan pada makanan kaleng khusus yang tidak mengandung susu atau produk yang mengandung susu.
Saat memilih produk makanan khusus untuk bayi, Anda harus membaca labelnya dengan cermat. Produk yang mengandung tidak lebih dari 5 mg galaktosa per 100 g dianggap aman untuk bayi.Jika terdapat lebih dari 20 mg galaktosa, dilarang keras memberikan produk kepada anak-anak.
Kecukupan pengobatan untuk galaktosemia diperiksa dengan pemeriksaan kontrol sekali dalam seperempat. Efektivitas pengobatan gejala patologi yang menyertainya juga diuji.
Penting! Perhatian khusus diberikan pada pemilihan obat untuk pasien tersebut. Beberapa obat mengandung laktosa dalam komposisinya, sementara yang lain dapat menghambat pembuangan produk metabolisme dari hati, yang umumnya memperburuk gejala kelainan bawaan dan tidak dapat diterima dalam kerangka pengobatan yang kompeten.Pencegahan galaktosemia

Tindakan pencegahan khusus untuk penyakit ini belum dikembangkan. Jika orang tua diketahui memiliki kecenderungan terhadap patologi, disarankan untuk merencanakan konsepsi dengan konsultasi tambahan dengan ahli genetika. Selama kehamilan, Anda juga harus menjalani serangkaian pemeriksaan (biopsi vili korionik pada minggu ke 10-12 dan tes cairan ketuban pada minggu ke 15-18) untuk mendeteksi mutasi GALT dan GALE pada gen.
Meskipun pengobatan gejala patologi telah dikembangkan dan cukup efektif, prospek jangka panjang perkembangan anak sulit diprediksi. Dengan demikian, indikator perkembangan fisik pasien akan lebih rendah dibandingkan teman sebayanya, kemungkinan besar akan terjadi gangguan bicara dan koordinasi, peningkatan kerapuhan tulang, dan pada anak perempuan - disfungsi ovarium.
Sejak usia 5 tahun, seorang anak mungkin dianjurkan untuk mengonsumsi vitamin dan obat yang mengandung ATP untuk mencegah perkembangan penyakit. Sejak usia 12 tahun, anak perempuan diberi resep terapi hormonal untuk mengkompensasi disfungsi ovarium.
Galaktosemia adalah penyakit genetik yang cukup langka. Mekanisme perkembangan patologi belum dipelajari secara menyeluruh, namun keseriusan masalah telah menyebabkan pengembangan sejumlah teknik diagnostik yang efektif. Skrining neonatal, yang diwajibkan di sejumlah negara, memungkinkan penyakit dideteksi tepat waktu dan mengambil tindakan. Meskipun pengobatan penyakit yang ditentukan secara genetik belum sepenuhnya dikembangkan, terapi membantu menghilangkan gejala penyakit yang parah dan perkembangannya. Tugas orang tua dalam hal ini adalah mendiagnosis patologi sedini mungkin dan mengikuti petunjuk dokter, sehingga membantu anak mengatasi kelainan bawaan tersebut.
Apa itu galaktosemia - tonton videonya:
