
What is galactosemia and why does it occur in a child? Symptoms, diagnosis of the disease. Treatment of galactosemia in newborns and available preventive measures.
The content of the article:- What is galactosemia
- Developmental symptoms
- Diagnostics
- How to treat galactosemia
- Prevention
Galactosemia is a rare but still common hereditary abnormality associated with metabolism. Enzymopathy is associated with dysfunction of carbohydrate metabolism, as a result of which the body accumulates galactose and its derivatives. Excessive amounts of galactose lead to the development of a clinical picture. In the case of late diagnosis of galactosemia, the symptoms become pronounced, and the prognosis for the normalization of the patient’s condition is bleak. Couples with a predisposition to the disease need to know about the types of galactosemia, methods of diagnosis, treatment and prevention, and also think about family planning. Timely measures taken significantly reduce the risk of complications in the newborn.
What is galactosemia?
Before talking about recommendations for galactosemia, it is necessary to understand the role of galactose in carbohydrate metabolism. This simple sugar enters the human body as part of lactose, a disaccharide the second component of which is glucose. Normally, the monosaccharide is used as “energy” resources for cells and complex compounds, “building” materials of nerve tissue, membranes, and nerve endings.
In a healthy person, lactose is broken down into its components in the digestive tract and gradually processed. Thus, at the first stage of processing, galactose 1-phosphate is obtained under the action of a special enzyme galactokinase and ATP. At the next stage, as a result of exchange under the action of enzymes, glucose 1-phosphate and uridine phosphate galactose are obtained. Monosaccharides are much more easily absorbed by the intestines during digestion, which is very important for newborns, since milk is the basis of their diet.
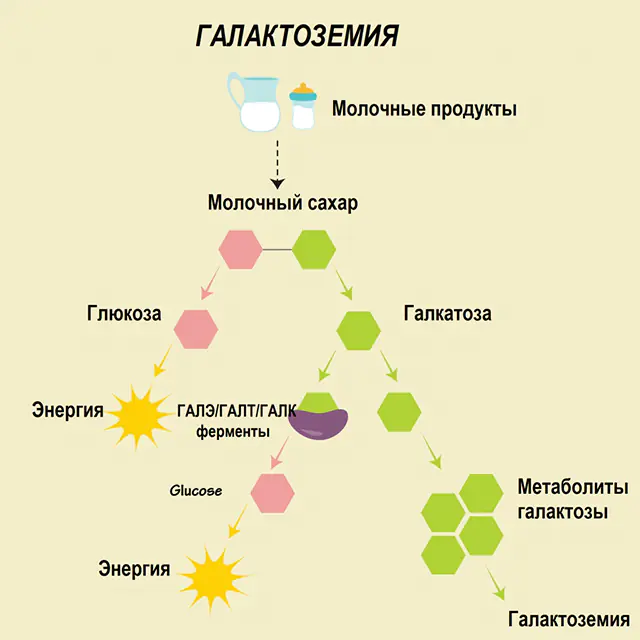
Three enzymes are involved in the processing processes - GALA (galactokinase), GALT (galactose 1-phosphate uridiyl transferase), GALE (epimerase). Deficient production of at least one of those enzymes at the first or second stage of carbohydrate metabolism leads to the accumulation of metabolites, intoxication of the body, and the subsequent manifestation of symptoms of galactosemia.
Depending on the defective enzyme, the corresponding types of galactosemia are distinguished:
- GALT violation- the first type occurs in one case per 40 thousand newborns; analysis for galactosemia will show reduced enzyme activity;
- GALC pathology- the second type occurs in one child out of 500 thousand, while the activity of the basic enzyme in erythrocytes is within the normal range, but the symptoms of the disease are pronounced;
- GALE— the third type was recorded the least (one case per million people) and is characterized by mild symptoms.
The most common type of disease (GALT) is divided into forms, depending on the genetics of galactosemia:
- The failure is characteristic of the stage of transformation of aspartic acid amide into aspartate - the pathology is also called Duarte's sign (Duarte). It is noteworthy that a person with such an anomaly will feel absolutely healthy, although the activity of the GALT enzyme is reduced by 25%-50%.
- Failure of the glutamine-arginine transition is most often recorded in Caucasians; enzyme activity is only 10% of the norm, which provokes a severe course of the disease.
- A disruption of the arginine-tryptophan transition occurs when the enzyme is completely inactive, an extremely severe form of the disease.
- Disruption of the lysine-asparagine transformation process occurs quite often.
- Failure of serine-leucine fermentation was recorded only in representatives of the Negroid race and is associated with insufficient enzyme activity in the liver (at a level of no more than 10% of the generally accepted norm).
The GALT enzyme is present in the body in excessive quantities, so a slight decrease in its activity does not always manifest itself as clinical symptoms. Only when GALT activity drops by 50 percent or more will the first signs of the disease appear.
Intoxication of the body with galactose and metabolites is dangerous for the development of complications in the long term: impaired motor function, speech, reproductive functions, especially in girls (despite the fact that the frequency of diseases in boys and girls is the same), growth retardation and other developmental dysfunctions.
Note! The pathogenesis of the disease has not yet been fully studied. However, it has already been established that the accumulation of galactose in the body is dangerous not only because of its toxic dosage, but also because of its inhibitory effect on other enzymes. Without proper treatment and control, the patient may develop hypoglycemic syndrome.As already noted, the pathology is a congenital anomaly. The type of inheritance of galactosemia is autosomal recessive, that is, the child receives defective genes from the parents. The mutated gene can be found in different alleles (forms of the condition) - defective and normal, when the defect was transmitted from only one parent. In this case, the disease manifests itself to a lesser extent. But if defective alleles have been passed on from both the father and mother in accordance with the type of inheritance of galactosemia, then the first signs of the disease will appear within a few days after the birth of the baby. It is noteworthy that if a disease is diagnosed in one child in a family, the likelihood of the pathology occurring in the next child from the same parents increases by 25%.
Insufficient production of enzymes leads to the accumulation of galactose in the body, and as a result, signs of galactosemia appear and the functions of the excretory system are disrupted. Without treatment within a few months of life, a person dies from liver failure or infections affecting the body.
It is important that the main causes of galactosemia (hereditary component) can be established even before the birth of the child, but doctors do not exclude the influence of other indicators on the development of the pathology:
- other gene mutations;
- affected tissue of the liver and central nervous system;
- accumulation of fluid in the lens of the eye, blurred vision.
And yet, if a patient is diagnosed with galactosemia, genetics plays a key role. Concomitant factors are important only in heterozygous patients, that is, those in whom the defective gene was transmitted from only one parent. In homozygous patients, the pathology itself is severe and requires highly specialized treatment.
Symptoms of the development of galactosemia
According to the severity of the manifestations of signs of galactosemia, three degrees of the disease are distinguished:
- light- usually detected accidentally due to milk intolerance;
- average- the first signs appear only after drinking milk;
- heavy— the symptoms of the pathology are superimposed on other malfunctions in the body (fluid accumulation in the abdominal cavity, sepsis).
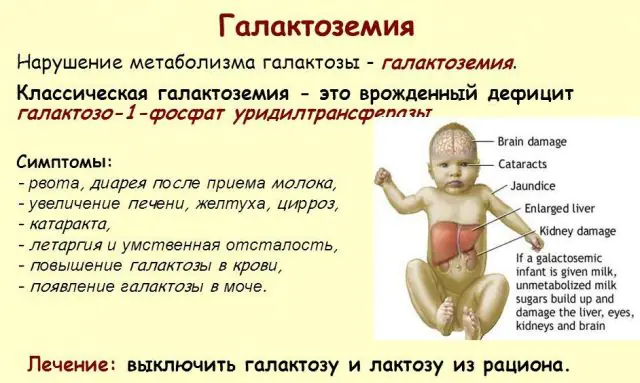
Classic GALT pathology manifests itself in its most severe form. The first signs of the disease will become visible after the birth of the child and his first feeding, since milk constitutes the main diet of the newborn. After feeding, he experiences vomiting, frequent bowel movements, and increased drowsiness against the background of constant lethargy and muscle hypotension. Despite regular breastfeeding, there is no weight gain. Digestive disorders are one of the first signs of dysfunction.
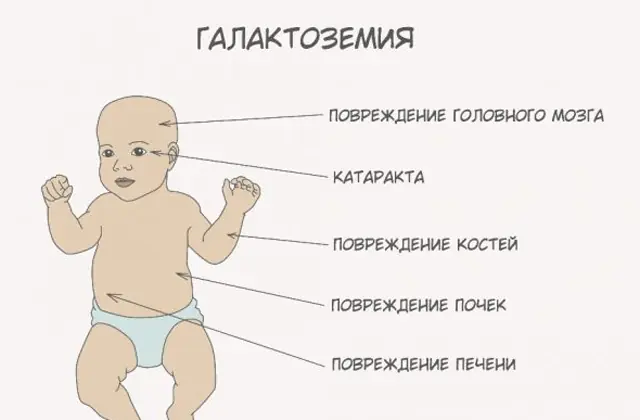
As the body becomes intoxicated, signs of liver damage become pronounced - jaundice, enlargement of the liver. Within a few weeks, the child develops cataracts, and after a few months, due to intoxication of the nerve tissues, disturbances in psychomotor functions are noted. The activity of the renal tubules is disrupted, as a result of which reducing sugar appears in the urine, and decreased blood clotting is noted.
Without the necessary therapy, the symptoms worsen as the baby’s body becomes intoxicated, and the risk to normal development and even human life increases. 20-30% of children, if treatment is refused or the disease is diagnosed late, die from sepsis, which develops due to leukocytotic activity. Those who survive suffer from kidney failure and psychomotor development problems.
In many ways, the degree of manifestation of the pathology depends on the hereditary cause. Galactosemia manifests itself as GALT deficiency occurs. Thus, Duhart's sign at first can only reveal itself as prolonged jaundice (up to 2 months after birth), cataracts and liver dysfunction are diagnosed a little later. But if the activity of the GALT enzyme is at least 50%, clinical symptoms may not appear.
The lack of GALA enzyme is not so pronounced. The main symptom of the disease is progressive cataracts, and monosaccharides may also appear in the urine over time. At the same time, the baby’s weight loss is insignificant, and psychomotor skills develop within the normal range.
GALE deficiency is initially extremely rare, but the symptoms are still divided into two forms: benign, when the lack of the enzyme affects blood cells, and generalized, when the deficiency affects all tissues of the body. In the first case, only an analysis for galactosemia will help detect the disease in the early stages; only with time will dysfunction of psychomotor skills and speech development be noted. The second form is similar in its symptoms to GALT deficiency, but in this case there is also an enlargement of the spleen. Even with the treatment of a severe form of GALE disease, over time the child experiences delayed psychomotor development, deafness, and vision problems.
Diagnosis of galactosemia
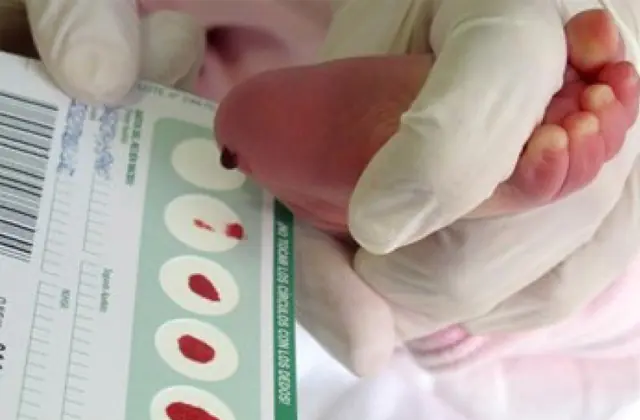
Neonatal screening, introduced in a number of countries, makes it possible to identify up to 50 congenital diseases in the early stages, including insufficient production of enzymes that are actively involved in metabolism. If for any reason screening is not carried out, the disease is identified based on aggregate characteristics.
So, if galactosemia is suspected, doctor’s recommendations are made only after a detailed diagnosis. A small patient must undergo:
- general examination - whether signs of jaundice are present;
- general signs are assessed - the quantity and quality of bowel movements, the presence or absence of muscle hypotonicity, problems with psychomotor skills;
- laboratory tests - blood test for biochemistry, general blood test, urine test for sugar and protein;
- additional examination of the condition of the liver and kidneys.
DNA diagnostics are mandatory to detect GALT mutations. If the child exhibits symptoms of the disease, and the activity of GALT enzymes according to test results is normal, DNA diagnostics is carried out for the GALE enzyme.
Doctors may prescribe a number of additional tests to differentiate the diagnosis. Exclusive diagnosis is necessary to confirm or refute the presence of diabetes mellitus and other pathologies associated with high sugar levels, as well as diseases that cause liver enlargement.
How to treat galactosemia?

Once neonatal screening and other examinations have confirmed the presence of galactosemia, treatment is prescribed. It is necessary to understand that since the disease is caused by a genetic predisposition, a complete cure at this stage of medical development is impossible. Therapy is aimed at reducing the clinical manifestations of the pathology, minimizing the progression of the disease and the development of complications.
The main treatment method is proper nutrition for galactosemia. All milk-containing products, even bread, sweets, sausages and others, are completely excluded from the patient’s diet. Products of plant and animal origin containing oligosaccharides are also removed: legumes, soy, spinach, chocolate, nuts, liver, brains, eggs, and derivatives of these products. Strict dietary therapy is the only way to prevent intoxication of the body.
For newborns, nutrition for galactosemia is also specialized. Breast milk is gradually completely eliminated from the diet and replaced with lactose-free formulas and formulations based on soy protein isolate. The volume of nutritional ingredients in this case should correspond to the standards for a healthy child. If a baby exhibits symptoms of an allergy to soy protein isolate, mixtures based on casein hydrolysates are selected. The first intake of food mixture should be 1/10 of the child’s daily nutritional requirement. Within 5-7 days, breast milk is completely eliminated from the diet.
The first complementary foods for children with pathology are prescribed from 4 months. The diet is expanded with fruit juices (apple, pear and others). After half a month, fruit purees are introduced. Up to 1 year, the volume of juice and puree consumed per day increases to 30-50 ml. The first vegetable puree in water (except legumes) is introduced at 5 months. From 5.5 months, a child can be given dairy-free commercial cereals (buckwheat, rice, corn). The baby can try meat products from the age of 6 months; preference should be given to specialized canned foods that do not contain milk or milk-containing products.
When choosing specialized food products for infants, you must carefully read the labels. Products that contain no more than 5 mg of galactose per 100 g are considered safe for a baby. If there is more than 20 mg of galactose, giving the product to a child is strictly prohibited.
The adequacy of treatment for galactosemia is checked by a control examination once a quarter. The effectiveness of treatment of accompanying symptoms of pathology is also tested.
Important! Special attention is paid to the selection of medications for such patients. Some drugs contain lactose in their composition, while others can inhibit the removal of metabolic products from the liver, which generally aggravates the symptoms of the congenital anomaly and is unacceptable within the framework of competent treatment.Prevention of galactosemia

Specialized preventive measures for the disease have not yet been developed. If parents have been found to have a predisposition to the pathology, it is recommended to plan conception with additional consultations with a geneticist. During pregnancy, you should also undergo a series of examinations (chorionic villus biopsy at 10-12 weeks and amniotic fluid testing at 15-18 weeks) to detect GALT and GALE mutations in genes.
Despite the developed quite effective symptomatic treatment of the pathology, the long-term prospects for the child’s development are difficult to predict. Thus, the patient’s physical development indicators will be lower than those of peers, speech and coordination disorders are likely to develop, increased bone fragility, and in girls - ovarian dysfunction.
From the age of 5, a child may be recommended to take vitamins and ATP-containing drugs to prevent the progression of the disease. From the age of 12, girls are prescribed hormonal therapy to compensate for ovarian dysfunction.
Galactosemia is a fairly rare genetically determined disease. The mechanism of development of the pathology has not yet been thoroughly studied, but the seriousness of the problem has led to the development of a number of effective diagnostic techniques. Neonatal screening, mandatory in a number of countries, makes it possible to detect the disease in time and take action. And although the treatment of genetically determined diseases has not been fully developed to date, therapy helps to get rid of severe symptoms of the disease and its progress. The task of the parents in this case is to diagnose the pathology as early as possible and follow the doctor’s instructions, thus helping the child cope with the congenital anomaly.
What is galactosemia - watch the video:
