
Hvad er galaktosæmi, og hvorfor opstår det hos et barn? Symptomer, diagnose af sygdommen. Behandling af galaktosæmi hos nyfødte og tilgængelige forebyggende foranstaltninger.
Artiklens indhold:- Hvad er galaktosæmi
- Udviklingssymptomer
- Diagnostik
- Hvordan man behandler galaktosæmi
- Forebyggelse
Galaktosæmi er en sjælden, men stadig almindelig arvelig abnormitet forbundet med metabolisme. Enzymopati er forbundet med dysfunktion af kulhydratmetabolisme, som et resultat af hvilken kroppen akkumulerer galactose og dets derivater. For store mængder galactose fører til udviklingen af et klinisk billede. I tilfælde af sen diagnose af galaktosæmi bliver symptomerne udtalte, og prognosen for normalisering af patientens tilstand er dyster. Par med en disposition for sygdommen har brug for at vide om typerne af galaktosæmi, metoder til diagnose, behandling og forebyggelse og også tænke på familieplanlægning. Rettidig truffet foranstaltninger reducerer betydeligt risikoen for komplikationer hos den nyfødte.
Hvad er galaktosæmi?
Før man taler om anbefalinger til galaktosæmi, er det nødvendigt at forstå galactoses rolle i kulhydratmetabolismen. Dette simple sukker kommer ind i den menneskelige krop som en del af laktose, et disaccharid, hvis anden komponent er glucose. Normalt bruges monosaccharidet som "energi" ressourcer til celler og komplekse forbindelser, "bygge" materialer af nervevæv, membraner og nerveender.
Hos en sund person nedbrydes laktose i dets komponenter i fordøjelseskanalen og behandles gradvist. Således opnås galactose 1-phosphat i det første trin af forarbejdningen under påvirkning af et specielt enzym galactokinase og ATP. På det næste trin, som et resultat af udveksling under påvirkning af enzymer, opnås glucose 1-phosphat og uridinphosphat galactose. Monosaccharider absorberes meget lettere af tarmene under fordøjelsen, hvilket er meget vigtigt for nyfødte, da mælk er grundlaget for deres kost.
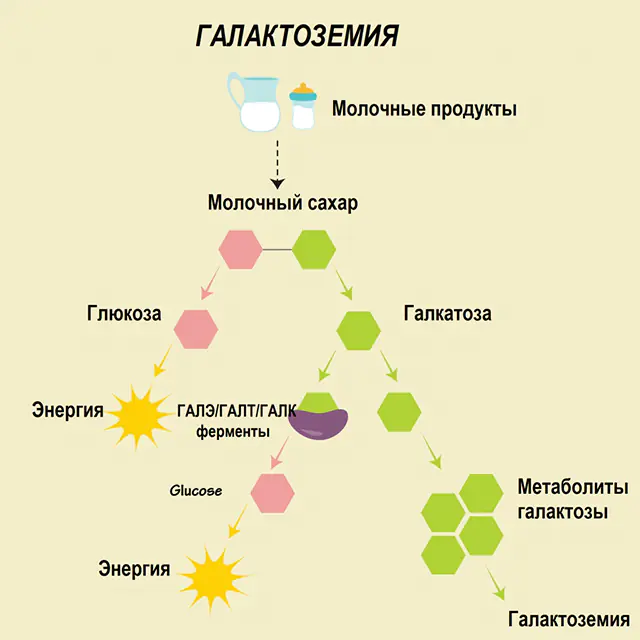
Tre enzymer er involveret i procesprocesserne - GALA (galactokinase), GALT (galactose 1-phosphat uridiyl transferase), GALE (epimerase). Mangelfuld produktion af mindst et af disse enzymer i det første eller andet trin af kulhydratmetabolismen fører til ophobning af metabolitter, forgiftning af kroppen og den efterfølgende manifestation af symptomer på galaktosæmi.
Afhængigt af det defekte enzym skelnes de tilsvarende typer galaktosæmi:
- GALT overtrædelse- den første type forekommer i ét tilfælde pr. 40 tusind nyfødte; analyse for galaktosæmi vil vise reduceret enzymaktivitet;
- GALC patologi- den anden type forekommer hos et barn ud af 500 tusind, mens aktiviteten af det grundlæggende enzym i erytrocytter er inden for det normale område, men symptomerne på sygdommen er udtalt;
- GALE— den tredje type blev registreret mindst (et tilfælde pr. million mennesker) og er karakteriseret ved milde symptomer.
Den mest almindelige type sygdom (GALT) er opdelt i former, afhængigt af genetikken for galaktosæmi:
- Svigtet er karakteristisk for omdannelsesstadiet af asparaginsyreamid til aspartat - patologien kaldes også Duartes tegn (Duarte). Det er bemærkelsesværdigt, at en person med en sådan anomali vil føle sig absolut sund, selvom aktiviteten af GALT-enzymet reduceres med 25% -50%.
- Svigt af glutamin-arginin-overgangen er oftest registreret hos kaukasiere; enzymaktivitet er kun 10% af normen, hvilket fremkalder et alvorligt sygdomsforløb.
- En forstyrrelse af arginin-tryptofan-overgangen opstår, når enzymet er fuldstændig inaktivt, en ekstremt alvorlig form for sygdommen.
- Afbrydelse af lysin-asparagin-transformationsprocessen forekommer ret ofte.
- Svigt af serin-leucin-fermentering blev kun registreret hos repræsentanter for den negroide race og er forbundet med utilstrækkelig enzymaktivitet i leveren (på et niveau på ikke mere end 10% af den generelt accepterede norm).
GALT-enzymet er til stede i kroppen i for store mængder, så et lille fald i dets aktivitet viser sig ikke altid som kliniske symptomer. Først når GALT-aktiviteten falder med 50 procent eller mere, vil de første tegn på sygdommen vise sig.
Forgiftning af kroppen med galactose og metabolitter er farligt for udviklingen af komplikationer på lang sigt: nedsat motorisk funktion, tale, reproduktive funktioner, især hos piger (på trods af, at hyppigheden af sygdomme hos drenge og piger er den samme), væksthæmning og andre udviklingsforstyrrelser.
Bemærk! Sygdommens patogenes er endnu ikke fuldt ud undersøgt. Det er imidlertid allerede blevet fastslået, at ophobning af galactose i kroppen er farlig ikke kun på grund af dens toksiske dosering, men også på grund af dens hæmmende virkning på andre enzymer. Uden ordentlig behandling og kontrol kan patienten udvikle hypoglykæmisk syndrom.Som allerede nævnt er patologien en medfødt anomali. Arten af nedarvning af galaktosæmi er autosomal recessiv, det vil sige, at barnet modtager defekte gener fra forældrene. Det muterede gen kan findes i forskellige alleler (tilstandens former) - defekte og normale, når defekten kun blev overført fra den ene forælder. I dette tilfælde manifesterer sygdommen sig i mindre grad. Men hvis defekte alleler er blevet videregivet fra både faderen og moderen i overensstemmelse med arten af arv af galaktosæmi, så vil de første tegn på sygdommen vise sig inden for et par dage efter fødslen af barnet. Det er bemærkelsesværdigt, at hvis en sygdom diagnosticeres i et barn i en familie, stiger sandsynligheden for, at patologien opstår i det næste barn fra de samme forældre med 25%.
Utilstrækkelig produktion af enzymer fører til ophobning af galactose i kroppen, og som et resultat opstår tegn på galaktosæmi, og udskillelsessystemets funktioner forstyrres. Uden behandling inden for et par måneder af livet, dør en person af leversvigt eller infektioner, der påvirker kroppen.
Det er vigtigt, at hovedårsagerne til galaktosæmi (arvelig komponent) kan fastslås selv før barnets fødsel, men læger udelukker ikke indflydelsen af andre indikatorer på udviklingen af patologien:
- andre genmutationer;
- påvirket væv i leveren og centralnervesystemet;
- ophobning af væske i øjets linse, sløret syn.
Og alligevel, hvis en patient bliver diagnosticeret med galaktosæmi, spiller genetik en nøglerolle. Samtidige faktorer er kun vigtige hos heterozygote patienter, det vil sige dem, hvor det defekte gen kun blev overført fra én forælder. Hos homozygote patienter er selve patologien alvorlig og kræver højt specialiseret behandling.
Symptomer på udvikling af galaktosæmi
Ifølge sværhedsgraden af manifestationerne af tegn på galaktosæmi skelnes tre grader af sygdommen:
- lys- normalt opdaget ved et uheld på grund af mælkeintolerance;
- gennemsnit- de første tegn vises først efter at have drukket mælk;
- tung— symptomerne på patologien er overlejret på andre funktionsfejl i kroppen (væskeophobning i bughulen, sepsis).
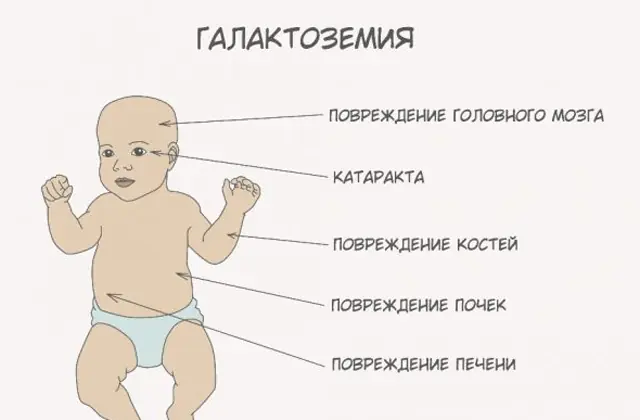
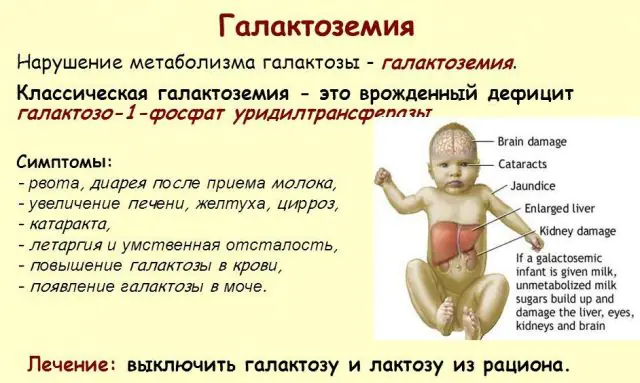
Klassisk GALT-patologi manifesterer sig i sin mest alvorlige form. De første tegn på sygdommen vil blive synlige efter barnets fødsel og hans første fodring, da mælk udgør den nyfødtes hoveddiæt. Efter fodring oplever han opkastning, hyppige afføringer og øget døsighed på baggrund af konstant sløvhed og muskelhypotension. På trods af regelmæssig amning er der ingen vægtøgning. Fordøjelsesforstyrrelser er et af de første tegn på dysfunktion.
Efterhånden som kroppen bliver beruset, bliver tegn på leverskade udtalt - gulsot, forstørrelse af leveren. Inden for et par uger udvikler barnet grå stær, og efter et par måneder, på grund af forgiftning af nervevævene, noteres forstyrrelser i psykomotoriske funktioner. Nyretubulis aktivitet forstyrres, som et resultat af hvilket reducerende sukker forekommer i urinen, og der noteres nedsat blodkoagulation.
Uden den nødvendige terapi forværres symptomerne, når babyens krop bliver beruset, og risikoen for normal udvikling og endda menneskeliv øges. 20-30 % af børn, hvis behandling afvises eller sygdommen diagnosticeres sent, dør af sepsis, som udvikler sig på grund af leukocytotisk aktivitet. De, der overlever, lider af nyresvigt og psykomotoriske udviklingsproblemer.
På mange måder afhænger graden af manifestation af patologien af den arvelige årsag. Galaktosæmi viser sig, når der opstår GALT-mangel. Således kan Duharts tegn i første omgang kun afsløre sig som langvarig gulsot (op til 2 måneder efter fødslen), grå stær og leverdysfunktion diagnosticeres lidt senere. Men hvis aktiviteten af GALT-enzymet er mindst 50 %, opstår der muligvis ikke kliniske symptomer.
Manglen på GALA-enzym er ikke så udtalt. Sygdommens hovedsymptom er fremadskridende grå stær, og monosakkarider kan også opstå i urinen over tid. Samtidig er barnets vægttab ubetydeligt, og psykomotoriske færdigheder udvikles inden for normalområdet.
GALE-mangel er i starten ekstremt sjælden, men symptomerne opdeles stadig i to former: godartet, når manglen på enzymet påvirker blodcellerne, og generaliseret, når manglen påvirker alle kroppens væv. I det første tilfælde vil kun en analyse for galaktosæmi hjælpe med at opdage sygdommen i de tidlige stadier; kun med tiden vil dysfunktion af psykomotoriske færdigheder og taleudvikling blive noteret. Den anden form ligner i sine symptomer GALT-mangel, men i dette tilfælde er der også en forstørrelse af milten. Selv ved behandling af en alvorlig form for GALE-sygdom oplever barnet over tid forsinket psykomotorisk udvikling, døvhed og synsproblemer.
Diagnose af galaktosæmi
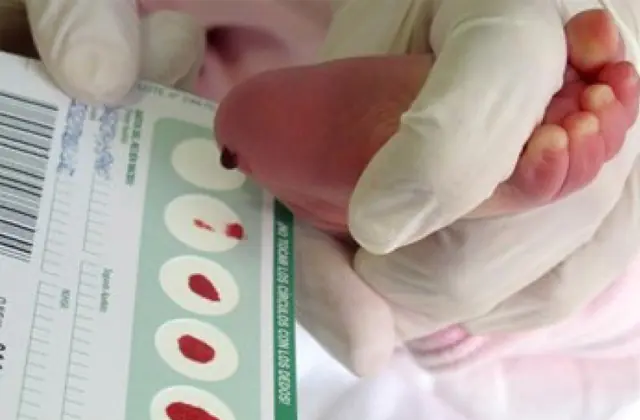
Neonatal screening, der er indført i en række lande, gør det muligt at identificere op til 50 medfødte sygdomme i de tidlige stadier, herunder utilstrækkelig produktion af enzymer, der er aktivt involveret i stofskiftet. Hvis der af en eller anden grund ikke udføres screening, identificeres sygdommen ud fra samlede karakteristika.
Så hvis der er mistanke om galaktosæmi, foretages lægens anbefalinger kun efter en detaljeret diagnose. En lille patient skal gennemgå:
- generel undersøgelse - om tegn på gulsot er til stede;
- generelle tegn vurderes - mængden og kvaliteten af afføring, tilstedeværelsen eller fraværet af muskelhypotoni, problemer med psykomotoriske færdigheder;
- laboratorieprøver - blodprøve for biokemi, generel blodprøve, urinprøve for sukker og protein;
- yderligere undersøgelse af lever- og nyrernes tilstand.
DNA-diagnostik er obligatorisk for at påvise GALT-mutationer. Hvis barnet udviser symptomer på sygdommen, og aktiviteten af GALT-enzymer ifølge testresultater er normal, udføres DNA-diagnostik for GALE-enzymet.
Læger kan ordinere en række yderligere tests for at differentiere diagnosen. Eksklusiv diagnose er nødvendig for at bekræfte eller afkræfte tilstedeværelsen af diabetes mellitus og andre patologier forbundet med høje sukkerniveauer samt sygdomme, der forårsager leverforstørrelse.
Hvordan behandler man galaktosæmi?

Når neonatal screening og andre undersøgelser har bekræftet tilstedeværelsen af galaktosæmi, ordineres behandling. Det er nødvendigt at forstå, at da sygdommen er forårsaget af en genetisk disposition, er en fuldstændig helbredelse på dette stadium af medicinsk udvikling umulig. Terapi er rettet mod at reducere de kliniske manifestationer af patologien, minimere sygdommens fremskridt og udviklingen af komplikationer.
Den vigtigste behandlingsmetode er korrekt ernæring til galaktosæmi. Alle mælkeholdige produkter, selv brød, slik, pølser og andre, er fuldstændig udelukket fra patientens kost. Produkter af vegetabilsk og animalsk oprindelse, der indeholder oligosaccharider, fjernes også: bælgfrugter, soja, spinat, chokolade, nødder, lever, hjerner, æg og derivater af disse produkter. Streng diætterapi er den eneste måde at forhindre forgiftning af kroppen på.
For nyfødte er ernæring til galaktosæmi også specialiseret. Modermælk fjernes gradvist fuldstændigt fra kosten og erstattes med laktosefri formler og formuleringer baseret på sojaproteinisolat. Mængden af ernæringsmæssige ingredienser i dette tilfælde skal svare til standarderne for et sundt barn. Hvis en baby udviser symptomer på allergi over for sojaproteinisolat, vælges blandinger baseret på kaseinhydrolysater. Det første indtag af madblanding bør være 1/10 af barnets daglige ernæringsbehov. Inden for 5-7 dage er modermælk fuldstændig elimineret fra kosten.
De første komplementære fødevarer til børn med patologi er ordineret fra 4 måneder. Kosten udvides med frugtjuice (æble, pære og andre). Efter en halv måned introduceres frugtpuréer. Op til 1 år stiger mængden af juice og puré forbrugt om dagen til 30-50 ml. Den første vegetabilske puré i vand (undtagen bælgfrugter) introduceres efter 5 måneder. Fra 5,5 måneder kan et barn få mælkefri kommercielle kornprodukter (boghvede, ris, majs). Babyen kan prøve kødprodukter fra 6-måneders alderen; der bør gives fortrinsret til specialkonserves, der ikke indeholder mælk eller mælkeholdige produkter.
Når du vælger specialfødevarer til spædbørn, skal du omhyggeligt læse etiketterne. Produkter, der ikke indeholder mere end 5 mg galactose pr. 100 g, anses for at være sikre for en baby. Hvis der er mere end 20 mg galactose, er det strengt forbudt at give produktet til et barn.
Behandlingens tilstrækkelighed for galaktosæmi kontrolleres ved en kontrolundersøgelse en gang i kvartalet. Effektiviteten af behandlingen af ledsagende symptomer på patologi testes også.
Vigtig! Der lægges særlig vægt på udvælgelsen af medicin til sådanne patienter. Nogle lægemidler indeholder laktose i deres sammensætning, mens andre kan hæmme fjernelse af metaboliske produkter fra leveren, hvilket generelt forværrer symptomerne på den medfødte anomali og er uacceptabel inden for rammerne af kompetent behandling.Forebyggelse af galaktosæmi

Der er endnu ikke udviklet specialiserede forebyggende foranstaltninger for sygdommen. Hvis forældre har vist sig at have en disposition for patologien, anbefales det at planlægge undfangelse med yderligere konsultationer med en genetiker. Under graviditeten bør du også gennemgå en række undersøgelser (chorionic villus biopsi ved 10-12 uger og fostervandstest ved 15-18 uger) for at påvise GALT og GALE mutationer i gener.
På trods af den udviklede ret effektive symptomatisk behandling af patologien er de langsigtede udsigter for barnets udvikling svære at forudsige. Således vil patientens fysiske udviklingsindikatorer være lavere end jævnaldrende, tale- og koordinationsforstyrrelser vil sandsynligvis udvikle sig, øget knogleskørhed og hos piger - ovariedysfunktion.
Fra 5 års alderen kan et barn blive anbefalet at tage vitaminer og ATP-holdige lægemidler for at forhindre udviklingen af sygdommen. Fra de er 12 år får piger ordineret hormonbehandling for at kompensere for ovariedysfunktion.
Galaktosæmi er en ret sjælden genetisk betinget sygdom. Mekanismen for udvikling af patologien er endnu ikke blevet grundigt undersøgt, men problemets alvor har ført til udviklingen af en række effektive diagnostiske teknikker. Neonatal screening, som er obligatorisk i en række lande, gør det muligt at opdage sygdommen i tide og gribe ind. Og selvom behandlingen af genetisk betingede sygdomme ikke er fuldt udviklet til dato, hjælper terapi med at slippe af med alvorlige symptomer på sygdommen og dens fremskridt. Forældrenes opgave i dette tilfælde er at diagnosticere patologien så tidligt som muligt og følge lægens instruktioner og dermed hjælpe barnet med at klare den medfødte anomali.
Hvad er galaktosæmi - se videoen:
