
O que é galactosemia e por que ocorre em crianças? Sintomas, diagnóstico da doença. Tratamento da galactosemia em recém-nascidos e medidas preventivas disponíveis.
O conteúdo do artigo:- O que é galactosemia
- Sintomas de desenvolvimento
- Diagnóstico
- Como tratar a galactosemia
- Prevenção
A galactosemia é uma anormalidade hereditária rara, mas ainda comum, associada ao metabolismo. A enzimopatia está associada à disfunção do metabolismo dos carboidratos, como resultado do acúmulo de galactose e seus derivados no corpo. Quantidades excessivas de galactose levam ao desenvolvimento de um quadro clínico. No caso de diagnóstico tardio da galactosemia, os sintomas tornam-se pronunciados e o prognóstico para a normalização do quadro do paciente é sombrio. Casais com predisposição à doença precisam conhecer os tipos de galactosemia, métodos de diagnóstico, tratamento e prevenção, além de pensar no planejamento familiar. As medidas oportunas tomadas reduzem significativamente o risco de complicações no recém-nascido.
O que é galactosemia?
Antes de falar sobre recomendações para galactosemia, é necessário compreender o papel da galactose no metabolismo dos carboidratos. Este açúcar simples entra no corpo humano como parte da lactose, um dissacarídeo cujo segundo componente é a glicose. Normalmente, o monossacarídeo é usado como recurso “energético” para células e compostos complexos, materiais de “construção” de tecido nervoso, membranas e terminações nervosas.
Em uma pessoa saudável, a lactose é decomposta em seus componentes no trato digestivo e gradualmente processada. Assim, na primeira etapa do processamento, a galactose 1-fosfato é obtida sob a ação de uma enzima especial galactoquinase e ATP. Na etapa seguinte, como resultado da troca sob a ação de enzimas, obtêm-se a glicose 1-fosfato e a uridina fosfato galactose. Os monossacarídeos são muito mais facilmente absorvidos pelo intestino durante a digestão, o que é muito importante para o recém-nascido, já que o leite é a base de sua alimentação.
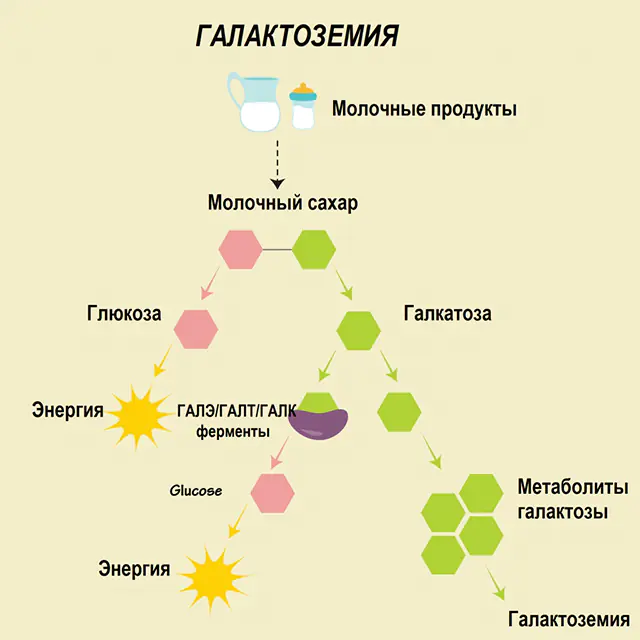
Três enzimas estão envolvidas nos processos de processamento - GALA (galactoquinase), GALT (galactose 1-fosfato uridiil transferase), GALE (epimerase). A produção deficiente de pelo menos uma dessas enzimas no primeiro ou segundo estágio do metabolismo dos carboidratos leva ao acúmulo de metabólitos, à intoxicação do corpo e à subsequente manifestação de sintomas de galactosemia.
Dependendo da enzima defeituosa, os tipos correspondentes de galactosemia são diferenciados:
- Violação GALT- o primeiro tipo ocorre em um caso a cada 40 mil recém-nascidos; a análise para galactosemia mostrará redução da atividade enzimática;
- Patologia GALC- o segundo tipo ocorre em uma criança em 500 mil, enquanto a atividade da enzima básica nos eritrócitos está dentro da normalidade, mas os sintomas da doença são pronunciados;
- GALE— o terceiro tipo foi o menos registado (um caso por milhão de pessoas) e caracteriza-se por sintomas ligeiros.
O tipo de doença mais comum (GALT) é dividido em formas, dependendo da genética da galactosemia:
- A falha é característica da fase de transformação da amida do ácido aspártico em aspartato - a patologia também é chamada de sinal de Duarte (Duarte). Vale ressaltar que uma pessoa com tal anomalia se sentirá absolutamente saudável, embora a atividade da enzima GALT seja reduzida em 25%-50%.
- A falha na transição glutamina-arginina é mais frequentemente registrada em caucasianos, a atividade enzimática é de apenas 10% do normal, o que provoca um curso grave da doença.
- Uma interrupção na transição arginina-triptofano ocorre quando a enzima está completamente inativa, uma forma extremamente grave da doença.
- A interrupção do processo de transformação lisina-asparagina ocorre com bastante frequência.
- A falha na fermentação da serina-leucina foi registrada apenas em representantes da raça negróide e está associada à atividade enzimática insuficiente no fígado (em um nível não superior a 10% da norma geralmente aceita).
A enzima GALT está presente no organismo em quantidades excessivas, pelo que uma ligeira diminuição da sua atividade nem sempre se manifesta como sintomas clínicos. Somente quando a atividade do GALT cair 50% ou mais é que aparecerão os primeiros sinais da doença.
A intoxicação do corpo com galactose e metabólitos é perigosa para o desenvolvimento de complicações a longo prazo: comprometimento da função motora, fala, funções reprodutivas, especialmente em meninas (apesar de a frequência de doenças em meninos e meninas ser a mesma), retardo de crescimento e outras disfunções de desenvolvimento.
Observação! A patogênese da doença ainda não foi totalmente estudada. No entanto, já foi estabelecido que o acúmulo de galactose no organismo é perigoso não só pela sua dosagem tóxica, mas também pelo seu efeito inibitório sobre outras enzimas. Sem tratamento e controle adequados, o paciente pode desenvolver síndrome hipoglicêmica.Como já foi observado, a patologia é uma anomalia congênita. O tipo de herança da galactosemia é autossômica recessiva, ou seja, a criança recebe genes defeituosos dos pais. O gene mutado pode ser encontrado em diferentes alelos (formas da doença) - defeituosos e normais, quando o defeito foi transmitido por apenas um dos pais. Nesse caso, a doença se manifesta em menor grau. Mas se alelos defeituosos foram transmitidos tanto pelo pai quanto pela mãe de acordo com o tipo de herança da galactosemia, os primeiros sinais da doença aparecerão alguns dias após o nascimento do bebê. Vale ressaltar que se uma doença for diagnosticada em uma criança da família, a probabilidade de a patologia ocorrer no próximo filho dos mesmos pais aumenta em 25%.
A produção insuficiente de enzimas leva ao acúmulo de galactose no corpo e, como resultado, aparecem sinais de galactosemia e as funções do sistema excretor são perturbadas. Sem tratamento dentro de alguns meses de vida, uma pessoa morre de insuficiência hepática ou infecções que afetam o corpo.
É importante que as principais causas da galactosemia (componente hereditário) possam ser estabelecidas antes mesmo do nascimento da criança, mas os médicos não excluem a influência de outros indicadores no desenvolvimento da patologia:
- outras mutações genéticas;
- tecido afetado do fígado e sistema nervoso central;
- acúmulo de líquido no cristalino do olho, visão turva.
E ainda assim, se um paciente for diagnosticado com galactosemia, a genética desempenha um papel fundamental. Fatores concomitantes são importantes apenas em pacientes heterozigotos, ou seja, aqueles nos quais o gene defeituoso foi transmitido por apenas um dos pais. Em pacientes homozigotos, a patologia em si é grave e requer tratamento altamente especializado.
Sintomas do desenvolvimento de galactosemia
De acordo com a gravidade das manifestações dos sinais de galactosemia, distinguem-se três graus da doença:
- luz- geralmente detectado acidentalmente devido à intolerância ao leite;
- média- os primeiros sinais aparecem somente após a ingestão de leite;
- pesado— os sintomas da patologia se sobrepõem a outras disfunções do corpo (acúmulo de líquidos na cavidade abdominal, sepse).
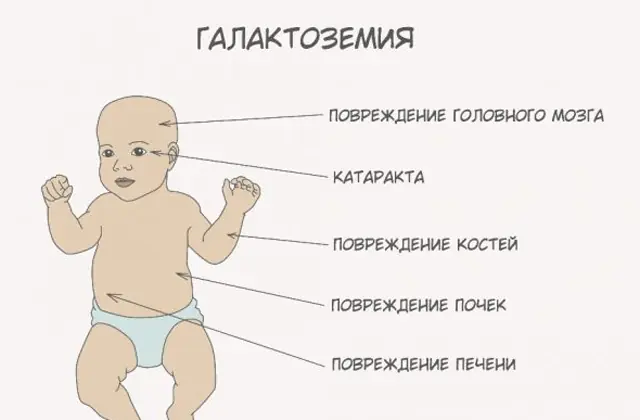
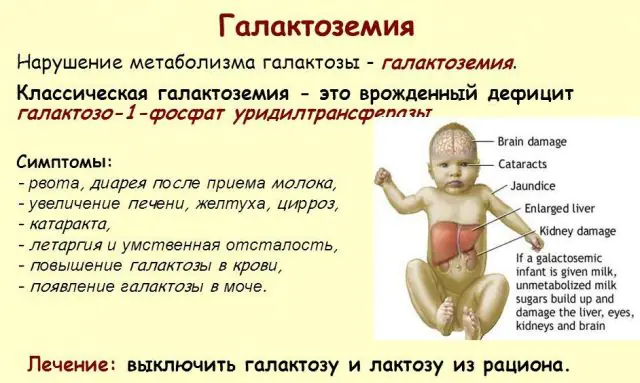
A patologia GALT clássica manifesta-se na sua forma mais grave. Os primeiros sinais da doença tornar-se-ão visíveis após o nascimento da criança e a sua primeira mamada, uma vez que o leite constitui a principal dieta do recém-nascido. Após a alimentação, ele apresenta vômitos, evacuações frequentes e aumento da sonolência num contexto de letargia constante e hipotensão muscular. Apesar da amamentação regular, não há ganho de peso. Os distúrbios digestivos são um dos primeiros sinais de disfunção.
À medida que o corpo fica intoxicado, os sinais de lesão hepática tornam-se pronunciados - icterícia, aumento do fígado. Dentro de algumas semanas, a criança desenvolve catarata e, após alguns meses, devido à intoxicação dos tecidos nervosos, são notados distúrbios nas funções psicomotoras. A atividade dos túbulos renais é interrompida, como resultado do aparecimento de açúcar redutor na urina e da diminuição da coagulação sanguínea.
Sem a terapia necessária, os sintomas pioram à medida que o corpo do bebê fica intoxicado e o risco ao desenvolvimento normal e até à vida humana aumenta. 20-30% das crianças, se o tratamento for recusado ou a doença for diagnosticada tardiamente, morrem de sepse, que se desenvolve devido à atividade leucocítica. Aqueles que sobrevivem sofrem de insuficiência renal e problemas de desenvolvimento psicomotor.
De muitas maneiras, o grau de manifestação da patologia depende da causa hereditária. A galactosemia se manifesta quando ocorre deficiência de GALT. Assim, o sinal de Duhart a princípio só pode se revelar como icterícia prolongada (até 2 meses após o nascimento), catarata e disfunção hepática são diagnosticadas um pouco mais tarde. Mas se a atividade da enzima GALT for de pelo menos 50%, os sintomas clínicos podem não aparecer.
A falta da enzima GALA não é tão pronunciada. O principal sintoma da doença é a catarata progressiva e, com o tempo, monossacarídeos também podem aparecer na urina. Ao mesmo tempo, a perda de peso do bebê é insignificante e as habilidades psicomotoras se desenvolvem dentro da normalidade.
A deficiência de GALE é inicialmente extremamente rara, mas os sintomas ainda se dividem em duas formas: benigna, quando a falta da enzima atinge as células sanguíneas, e generalizada, quando a deficiência atinge todos os tecidos do corpo. No primeiro caso, apenas uma análise de galactosemia ajudará a detectar a doença nos estágios iniciais, somente com o tempo será notada disfunção das habilidades psicomotoras e do desenvolvimento da fala. A segunda forma é semelhante em sintomas à deficiência de GALT, mas neste caso também ocorre aumento do baço. Mesmo com o tratamento de uma forma grave da doença GALE, com o tempo a criança apresenta atraso no desenvolvimento psicomotor, surdez e problemas de visão.
Diagnóstico de galactosemia
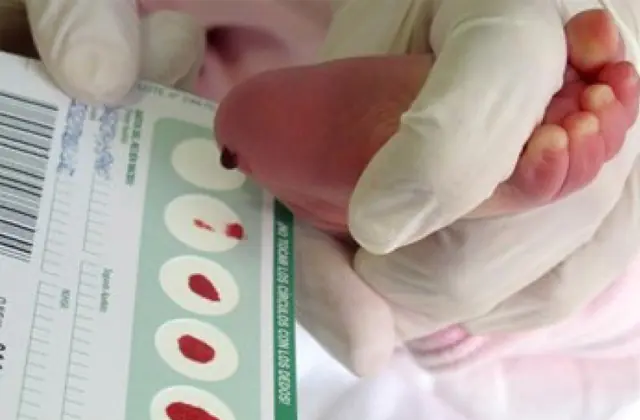
A triagem neonatal, introduzida em vários países, permite identificar até 50 doenças congênitas em estágios iniciais, incluindo a produção insuficiente de enzimas que estão ativamente envolvidas no metabolismo. Se por algum motivo não for realizado o rastreio, a doença é identificada com base em características agregadas.
Assim, se houver suspeita de galactosemia, as recomendações do médico só são feitas após um diagnóstico detalhado. Um paciente pequeno deve passar por:
- exame geral - se há sinais de icterícia;
- são avaliados sinais gerais - quantidade e qualidade das evacuações, presença ou ausência de hipotonicidade muscular, problemas psicomotores;
- exames laboratoriais - exame de sangue para bioquímica, exame de sangue geral, exame de urina para açúcar e proteínas;
- exame adicional da condição do fígado e dos rins.
O diagnóstico de DNA é obrigatório para detectar mutações GALT. Se a criança apresentar sintomas da doença e a atividade das enzimas GALT de acordo com os resultados dos exames for normal, é realizado o diagnóstico de DNA para a enzima GALE.
Os médicos podem prescrever vários testes adicionais para diferenciar o diagnóstico. O diagnóstico exclusivo é necessário para confirmar ou refutar a presença de diabetes mellitus e outras patologias associadas a níveis elevados de açúcar, bem como doenças que causam aumento do fígado.
Como tratar a galactosemia?

Uma vez que a triagem neonatal e outros exames confirmem a presença de galactosemia, o tratamento é prescrito. É preciso entender que como a doença é causada por uma predisposição genética, a cura completa nesta fase do desenvolvimento médico é impossível. A terapia visa reduzir as manifestações clínicas da patologia, minimizando a evolução da doença e o desenvolvimento de complicações.
O principal método de tratamento é a nutrição adequada para galactosemia. Todos os produtos lácteos, mesmo pães, doces, embutidos e outros, são totalmente excluídos da dieta do paciente. Também são retirados produtos de origem vegetal e animal contendo oligossacarídeos: legumes, soja, espinafre, chocolate, nozes, fígado, cérebro, ovos e derivados desses produtos. A terapia dietética rigorosa é a única maneira de prevenir a intoxicação do corpo.
Para os recém-nascidos, a nutrição para galactosemia também é especializada. O leite materno é gradativamente eliminado da dieta e substituído por fórmulas sem lactose e formulações à base de proteína isolada de soja. A quantidade de ingredientes nutricionais, neste caso, deve corresponder aos padrões para uma criança saudável. Se um bebê apresentar sintomas de alergia ao isolado de proteína de soja, são selecionadas misturas à base de hidrolisados de caseína. A primeira ingestão da mistura alimentar deve ser 1/10 das necessidades nutricionais diárias da criança. Dentro de 5 a 7 dias, o leite materno é completamente eliminado da dieta.
Os primeiros alimentos complementares para crianças com patologia são prescritos a partir dos 4 meses. A dieta é ampliada com sucos de frutas (maçã, pêra e outros). Depois de meio mês, são introduzidos purês de frutas. Até 1 ano, o volume de suco e purê consumido por dia aumenta para 30-50 ml. O primeiro purê de vegetais em água (exceto legumes) é introduzido aos 5 meses. A partir dos 5,5 meses, uma criança pode receber cereais comerciais sem laticínios (trigo sarraceno, arroz, milho). O bebê pode experimentar produtos cárneos a partir dos 6 meses, devendo-se dar preferência a alimentos enlatados especializados que não contenham leite ou produtos que contenham leite.
Ao escolher produtos alimentícios especializados para bebês, é necessário ler atentamente os rótulos. Produtos que não contenham mais de 5 mg de galactose por 100 g são considerados seguros para um bebê.Se houver mais de 20 mg de galactose, é estritamente proibido dar o produto a uma criança.
A adequação do tratamento da galactosemia é verificada por um exame de controle trimestral. A eficácia do tratamento dos sintomas acompanhantes da patologia também é testada.
Importante! Atenção especial é dada à seleção de medicamentos para esses pacientes. Alguns medicamentos contêm lactose em sua composição, enquanto outros podem inibir a eliminação de produtos metabólicos do fígado, o que geralmente agrava os sintomas da anomalia congênita e é inaceitável no âmbito de um tratamento competente.Prevenção da galactosemia

Medidas preventivas especializadas para a doença ainda não foram desenvolvidas. Se for constatado que os pais têm predisposição à patologia, é recomendável planejar a concepção com consultas adicionais com um geneticista. Durante a gravidez, você também deve passar por uma série de exames (biópsia de vilosidades coriônicas em 10-12 semanas e teste de líquido amniótico em 15-18 semanas) para detectar mutações GALT e GALE nos genes.
Apesar do tratamento sintomático bastante eficaz da patologia desenvolvido, as perspectivas de longo prazo para o desenvolvimento da criança são difíceis de prever. Assim, os indicadores de desenvolvimento físico do paciente serão inferiores aos dos pares, é provável que se desenvolvam distúrbios de fala e coordenação, aumento da fragilidade óssea e, nas meninas, disfunção ovariana.
A partir dos 5 anos de idade, pode ser recomendado que uma criança tome vitaminas e medicamentos contendo ATP para prevenir a progressão da doença. A partir dos 12 anos, as meninas recebem terapia hormonal para compensar a disfunção ovariana.
A galactosemia é uma doença geneticamente determinada bastante rara. O mecanismo de desenvolvimento da patologia ainda não foi completamente estudado, mas a gravidade do problema levou ao desenvolvimento de uma série de técnicas diagnósticas eficazes. A triagem neonatal, obrigatória em vários países, permite detectar a tempo a doença e agir. E embora o tratamento de doenças geneticamente determinadas não tenha sido totalmente desenvolvido até o momento, a terapia ajuda a eliminar os sintomas graves da doença e seu progresso. A tarefa dos pais, neste caso, é diagnosticar a patologia o mais cedo possível e seguir as orientações do médico, ajudando assim a criança a enfrentar a anomalia congênita.
O que é galactosemia - assista ao vídeo:
