
Galaktozemi nedir ve neden çocuklarda görülür? Belirtileri, hastalığın tanısı. Yenidoğanlarda galaktozeminin tedavisi ve mevcut önleyici tedbirler.
Makalenin içeriği:- Galaktozemi nedir
- Gelişimsel belirtiler
- Teşhis
- Galaktozemi nasıl tedavi edilir
- Önleme
Galaktozemi, metabolizmayla ilişkili nadir fakat yine de yaygın görülen kalıtsal bir anormalliktir. Enzimopati, vücudun galaktoz ve türevlerini biriktirmesinin bir sonucu olarak karbonhidrat metabolizmasının işlev bozukluğu ile ilişkilidir. Aşırı miktarda galaktoz klinik bir tablonun gelişmesine yol açar. Galaktozeminin geç teşhisi durumunda semptomlar belirginleşir ve hastanın durumunun normalleşmesine yönelik prognoz kasvetlidir. Hastalığa yatkınlığı olan çiftlerin galaktozemi türlerini, tanı, tedavi ve önleme yöntemlerini bilmeleri ve ayrıca aile planlaması hakkında düşünmeleri gerekir. Zamanında alınan önlemler yenidoğanda komplikasyon riskini önemli ölçüde azaltır.
Galaktozemi nedir?
Galaktozemiye yönelik önerilerden bahsetmeden önce, galaktozun karbonhidrat metabolizmasındaki rolünü anlamak gerekir. Bu basit şeker insan vücuduna, ikinci bileşeni glikoz olan bir disakkarit olan laktozun bir parçası olarak girer. Normalde monosakarit, hücreler ve karmaşık bileşikler için "enerji" kaynağı, sinir dokusunun, zarların ve sinir uçlarının "yapı malzemeleri" olarak kullanılır.
Sağlıklı bir insanda laktoz sindirim sisteminde bileşenlerine ayrılır ve yavaş yavaş işlenir. Böylece, işlemenin ilk aşamasında, özel bir galaktokinaz ve ATP enziminin etkisi altında galaktoz 1-fosfat elde edilir. Bir sonraki aşamada enzimlerin etkisi altındaki değişim sonucunda glikoz 1-fosfat ve üridin fosfat galaktoz elde edilir. Monosakkaritler, sindirim sırasında bağırsaklar tarafından çok daha kolay emilir ki bu, yenidoğanlar için çok önemlidir, çünkü süt onların diyetinin temelidir.
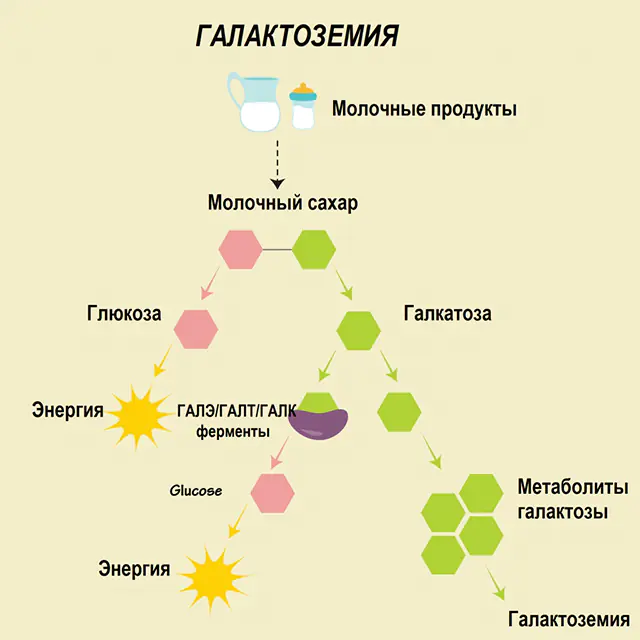
İşleme süreçlerinde üç enzim rol oynar - GALA (galaktokinaz), GALT (galaktoz 1-fosfat üridiil transferaz), GALE (epimeraz). Karbonhidrat metabolizmasının birinci veya ikinci aşamasında bu enzimlerden en az birinin yetersiz üretimi, metabolitlerin birikmesine, vücudun zehirlenmesine ve ardından galaktozemi semptomlarının ortaya çıkmasına neden olur.
Kusurlu enzime bağlı olarak, karşılık gelen galaktozemi türleri ayırt edilir:
- GALT ihlali- İlk tip 40 bin yenidoğanda bir vakada görülür; galaktozemi analizi enzim aktivitesinde azalma gösterecektir;
- GALC patolojisi- ikinci tip 500 bin çocuktan birinde görülür, eritrositlerdeki temel enzimin aktivitesi normal aralıktadır, ancak hastalığın belirtileri belirgindir;
- GALE— üçüncü tip en az kaydedilmiştir (milyon kişi başına bir vaka) ve hafif semptomlarla karakterizedir.
En yaygın hastalık türü (GALT), galaktozeminin genetiğine bağlı olarak formlara ayrılır:
- Başarısızlık, aspartik asit amidinin aspartata dönüşüm aşamasının karakteristiğidir - patolojiye Duarte işareti (Duarte) de denir. GALT enziminin aktivitesinin %25-%50 oranında azalmasına rağmen, böyle bir anomaliye sahip bir kişinin kendisini tamamen sağlıklı hissedeceği dikkat çekmektedir.
- Glutamin-arginin geçişinin başarısızlığı en sık Kafkasyalılarda kaydedilir, enzim aktivitesi normun sadece% 10'udur ve bu da hastalığın ciddi seyrini tetikler.
- Arginin-triptofan geçişinin bozulması, enzim tamamen inaktif olduğunda meydana gelir; bu, hastalığın son derece şiddetli bir şeklidir.
- Lizin-asparajin dönüşüm sürecinin bozulması oldukça sık meydana gelir.
- Serin-lösin fermantasyonunun başarısızlığı yalnızca Negroid ırkının temsilcilerinde kaydedildi ve karaciğerdeki yetersiz enzim aktivitesi ile ilişkilidir (genel kabul edilen normun% 10'undan fazla olmayan bir seviyede).
GALT enzimi vücutta aşırı miktarlarda bulunur, bu nedenle aktivitesinde hafif bir azalma her zaman klinik semptom olarak kendini göstermez. Ancak GALT aktivitesi yüzde 50 veya daha fazla düştüğünde hastalığın ilk belirtileri ortaya çıkacaktır.
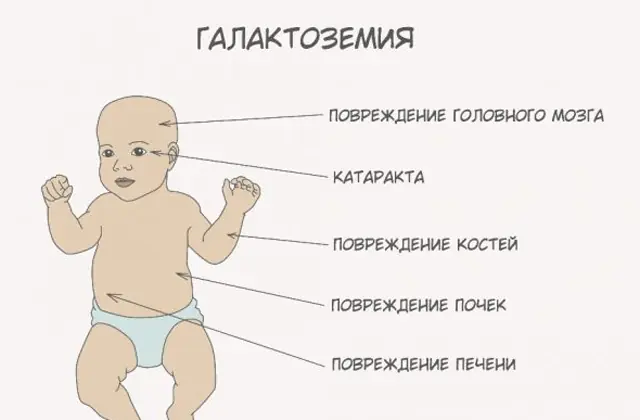
Vücudun galaktoz ve metabolitlerle zehirlenmesi, uzun vadede komplikasyonların gelişmesi açısından tehlikelidir: özellikle kızlarda motor fonksiyon, konuşma, üreme fonksiyonlarında bozulma (erkek ve kızlarda hastalık sıklığının aynı olmasına rağmen), Büyüme geriliği ve diğer gelişimsel işlev bozuklukları.
Not! Hastalığın patogenezi henüz tam olarak araştırılmamıştır. Ancak galaktozun vücutta birikmesinin sadece toksik dozajı nedeniyle değil aynı zamanda diğer enzimler üzerindeki engelleyici etkisi nedeniyle de tehlikeli olduğu zaten tespit edilmiştir. Uygun tedavi ve kontrol olmadan hastada hipoglisemik sendrom gelişebilir.Daha önce de belirtildiği gibi, patoloji doğuştan bir anomalidir. Galaktozeminin kalıtım türü otozomal resesiftir, yani çocuk ebeveynlerden kusurlu genler alır. Mutasyona uğramış gen farklı alellerde (durumun formları) bulunabilir - kusurlu ve normal, kusur yalnızca bir ebeveynden aktarıldığında. Bu durumda hastalık daha az kendini gösterir. Ancak galaktozeminin kalıtım türüne göre hem anneden hem de babadan kusurlu aleller aktarılmışsa, hastalığın ilk belirtileri bebeğin doğumundan sonraki birkaç gün içinde ortaya çıkacaktır. Ailede bir çocukta hastalık tanısı konulduğu takdirde aynı ebeveynden gelen bir sonraki çocukta da patolojinin ortaya çıkma ihtimalinin %25 oranında arttığı dikkat çekmektedir.
Yetersiz enzim üretimi vücutta galaktoz birikmesine yol açar ve bunun sonucunda galaktozemi belirtileri ortaya çıkar ve boşaltım sisteminin işlevleri bozulur. Tedavi edilmeyen kişi birkaç ay içinde karaciğer yetmezliğinden veya vücudu etkileyen enfeksiyonlardan ölür.
Galaktozeminin ana nedenlerinin (kalıtsal bileşen) çocuğun doğumundan önce bile belirlenebilmesi önemlidir, ancak doktorlar diğer göstergelerin patolojinin gelişimi üzerindeki etkisini dışlamaz:
- diğer gen mutasyonları;
- karaciğer ve merkezi sinir sisteminin etkilenen dokusu;
- Göz merceğinde sıvı birikmesi, bulanık görme.
Ancak bir hastaya galaktozemi teşhisi konursa genetik önemli bir rol oynar. Eşlik eden faktörler yalnızca heterozigot hastalarda, yani kusurlu genin yalnızca bir ebeveynden aktarıldığı hastalarda önemlidir. Homozigot hastalarda patolojinin kendisi ciddidir ve oldukça uzmanlaşmış tedavi gerektirir.
Galaktozemi gelişiminin belirtileri
Galaktozemi belirtilerinin belirtilerinin ciddiyetine göre, hastalığın üç derecesi ayırt edilir:
- ışık- genellikle süt intoleransı nedeniyle tesadüfen tespit edilir;
- ortalama- ilk belirtiler ancak süt içtikten sonra ortaya çıkar;
- ağır- patolojinin semptomları vücuttaki diğer arızaların üzerine bindirilir (karın boşluğunda sıvı birikmesi, sepsis).
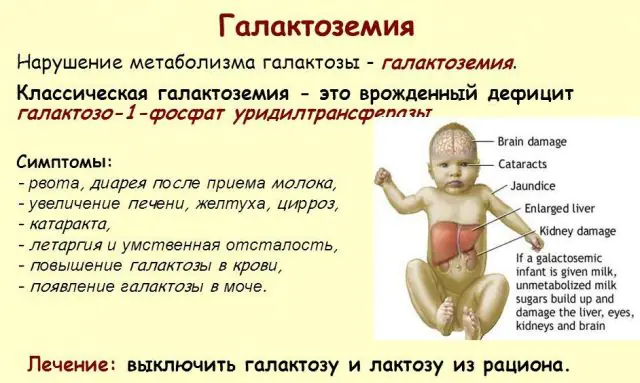
Klasik GALT patolojisi en şiddetli haliyle kendini gösterir. Süt, yenidoğanın ana diyetini oluşturduğundan, hastalığın ilk belirtileri çocuğun doğumundan ve ilk beslenmesinden sonra ortaya çıkacaktır. Beslendikten sonra kusma, sık bağırsak hareketleri ve sürekli uyuşukluk ve kas hipotansiyonunun arka planında artan uyuşukluk yaşar. Düzenli emzirmeye rağmen kilo alımı olmaz. Sindirim bozuklukları, işlev bozukluğunun ilk belirtilerinden biridir.
Vücut sarhoş oldukça, karaciğer hasarının belirtileri belirginleşir - sarılık, karaciğerin büyümesi. Birkaç hafta içinde çocukta katarakt gelişir ve birkaç ay sonra sinir dokularının zehirlenmesi nedeniyle psikomotor fonksiyonlarda rahatsızlıklar görülür. Böbrek tübüllerinin aktivitesi bozulur, bunun sonucunda idrarda şekerin azalması görülür ve kanın pıhtılaşmasında azalma görülür.
Gerekli tedavi yapılmazsa, bebeğin vücudu sarhoş hale geldikçe belirtiler daha da kötüleşir ve normal gelişim ve hatta insan hayatı için risk artar. Tedavinin reddedilmesi veya hastalığın geç teşhis edilmesi durumunda çocukların %20-30'u lökositotik aktiviteye bağlı gelişen sepsis nedeniyle ölmektedir. Hayatta kalanlar böbrek yetmezliği ve psikomotor gelişim sorunları yaşıyor.
Birçok yönden patolojinin tezahür derecesi kalıtsal nedene bağlıdır. Galaktozemi, GALT eksikliğinin ortaya çıkmasıyla kendini gösterir. Bu nedenle, Duhart'ın belirtisi ilk başta ancak uzun süreli sarılık (doğumdan sonra 2 aya kadar) olarak kendini gösterebilir, katarakt ve karaciğer fonksiyon bozuklukları bir süre sonra teşhis edilir. Ancak GALT enziminin aktivitesi en az %50 ise klinik belirtiler ortaya çıkmayabilir.
GALA enziminin eksikliği o kadar belirgin değildir. Hastalığın ana semptomu ilerleyici katarakttır ve zamanla idrarda monosakkaritler de ortaya çıkabilir. Aynı zamanda bebeğin kilo kaybı önemsizdir ve psikomotor becerileri normal aralıkta gelişir.
GALE eksikliği başlangıçta son derece nadirdir, ancak semptomlar yine de iki forma ayrılır: enzim eksikliği kan hücrelerini etkilediğinde iyi huyludur ve eksiklik vücudun tüm dokularını etkilediğinde genelleştirilmiştir. İlk durumda, yalnızca galaktozemi analizi hastalığın erken evrelerde tespit edilmesine yardımcı olacaktır, ancak zamanla psikomotor becerilerde ve konuşma gelişiminde işlev bozukluğu fark edilecektir. İkinci form, semptomları açısından GALT eksikliğine benzer, ancak bu durumda dalakta da bir genişleme vardır. GALE hastalığının ciddi bir formunun tedavisinde bile zamanla çocukta gecikmiş psikomotor gelişim, sağırlık ve görme sorunları ortaya çıkar.
Galaktozemi tanısı
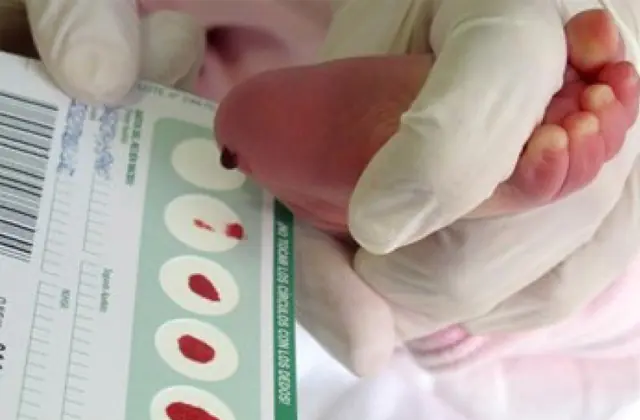
Birçok ülkede uygulamaya konan yenidoğan taraması, metabolizmada aktif olarak yer alan enzimlerin yetersiz üretimi de dahil olmak üzere 50'ye kadar konjenital hastalığın erken evrelerde tespit edilmesini mümkün kılmaktadır. Herhangi bir nedenle tarama yapılmazsa hastalık toplu özelliklere göre belirlenir.
Bu nedenle galaktozemiden şüpheleniliyorsa doktor tavsiyesi ancak detaylı tanı konulduktan sonra yapılır. Küçük bir hastanın geçmesi gerekir:
- genel muayene - sarılık belirtilerinin mevcut olup olmadığı;
- genel belirtiler değerlendirilir - bağırsak hareketlerinin miktarı ve kalitesi, kas hipotonisinin varlığı veya yokluğu, psikomotor becerilerle ilgili sorunlar;
- laboratuvar testleri - biyokimya için kan testi, genel kan testi, şeker ve protein için idrar testi;
- karaciğer ve böbreklerin durumunun ek muayenesi.
GALT mutasyonlarını tespit etmek için DNA teşhisi zorunludur. Çocukta hastalık belirtileri görülüyorsa ve test sonuçlarına göre GALT enzimlerinin aktivitesi normal ise GALE enzimi için DNA tanısı yapılır.
Doktorlar tanıyı ayırt etmek için bir dizi ek test önerebilir. Diyabetin ve yüksek şeker seviyeleriyle ilişkili diğer patolojilerin yanı sıra karaciğer büyümesine neden olan hastalıkların varlığını doğrulamak veya çürütmek için özel tanı gereklidir.
Galaktozemi nasıl tedavi edilir?

Yenidoğan taraması ve diğer muayeneler galaktozeminin varlığını doğruladıktan sonra tedavi reçete edilir. Hastalığın genetik yatkınlıktan kaynaklandığı için tıbbi gelişimin bu aşamasında tam bir tedavinin imkansız olduğunu anlamak gerekir. Terapi, patolojinin klinik belirtilerini azaltmayı, hastalığın ilerlemesini ve komplikasyonların gelişimini en aza indirmeyi amaçlamaktadır.
Galaktozeminin ana tedavi yöntemi doğru beslenmedir. Süt içeren tüm ürünler, hatta ekmek, tatlılar, sosisler ve diğerleri bile hastanın diyetinden tamamen çıkarılır. Oligosakkaritler içeren bitkisel ve hayvansal kökenli ürünler de çıkarılır: baklagiller, soya, ıspanak, çikolata, fındık, karaciğer, beyin, yumurta ve bu ürünlerin türevleri. Sıkı diyet tedavisi vücudun zehirlenmesini önlemenin tek yoludur.
Yeni doğanlar için galaktozemiye yönelik beslenme de uzmanlaşmıştır. Anne sütü yavaş yavaş diyetten tamamen çıkarılır ve yerini laktoz içermeyen formüller ve soya proteini izolatı bazlı formülasyonlar alır. Bu durumda besin bileşenlerinin hacmi sağlıklı bir çocuğun standartlarına uygun olmalıdır. Bir bebek soya proteini izolatına karşı alerji belirtileri gösteriyorsa kazein hidrolizatlarına dayalı karışımlar seçilir. Besin karışımının ilk alımı çocuğun günlük besin ihtiyacının 1/10'u kadar olmalıdır. 5-7 gün içerisinde anne sütü diyetten tamamen çıkarılır.
Patolojisi olan çocuklar için ilk tamamlayıcı gıdalar 4 aydan itibaren reçete edilir. Diyet meyve suları (elma, armut ve diğerleri) ile genişletilir. Yarım ay sonra meyve püreleri tanıtılır. 1 yıla kadar günde tüketilen meyve suyu ve püre miktarı 30-50 ml'ye çıkar. Suda ilk sebze püresi (baklagiller hariç) 5. ayda verilir. 5,5 aydan itibaren çocuğa süt içermeyen ticari tahıllar (karabuğday, pirinç, mısır) verilebilir. Bebek 6 aylıktan itibaren et ürünlerini deneyebilir, süt içermeyen veya süt içeren ürünler içermeyen özel konserve yiyecekler tercih edilmelidir.
Bebekler için özel gıda ürünleri seçerken etiketleri dikkatlice okumalısınız. 100 g'da 5 mg'dan fazla galaktoz içermeyen ürünler bebek için güvenli kabul edilir, 20 mg'dan fazla galaktoz varsa ürünün çocuğa verilmesi kesinlikle yasaktır.
Galaktozemi tedavisinin yeterliliği her üç ayda bir kontrol muayenesi ile kontrol edilir. Eşlik eden patoloji semptomlarının tedavisinin etkinliği de test edilmiştir.
Önemli! Bu tür hastalar için ilaç seçimine özellikle dikkat edilir. Bazı ilaçlar bileşimlerinde laktoz içerirken, diğerleri metabolik ürünlerin karaciğerden uzaklaştırılmasını engelleyebilir, bu da genellikle konjenital anomali semptomlarını ağırlaştırır ve yetkili tedavi çerçevesinde kabul edilemez.Galaktozeminin önlenmesi

Hastalık için özel önleyici tedbirler henüz geliştirilmemiştir. Ebeveynlerin patolojiye yatkınlığı olduğu tespit edilirse, bir genetikçiyle ek istişarelerle gebe kalmanın planlanması önerilir. Hamilelik sırasında ayrıca genlerdeki GALT ve GALE mutasyonlarını tespit etmek için bir dizi muayeneden (10-12. haftalarda koryon villus biyopsisi ve 15-18. haftalarda amniyotik sıvı testi) geçmelisiniz.
Patolojinin oldukça etkili semptomatik tedavisinin geliştirilmesine rağmen, çocuğun gelişimi için uzun vadeli beklentileri tahmin etmek zordur. Böylece hastanın fiziksel gelişim göstergeleri akranlarına göre daha düşük olacak, konuşma ve koordinasyon bozuklukları gelişebilecek, kemik kırılganlığı artacak ve kızlarda yumurtalık fonksiyon bozuklukları ortaya çıkabilecektir.
Hastalığın ilerlemesini önlemek için 5 yaşından itibaren çocuğa vitamin ve ATP içeren ilaçlar alması önerilebilir. 12 yaşından itibaren kızlara yumurtalık fonksiyon bozukluğunu telafi etmek için hormonal tedavi reçete edilir.
Galaktozemi oldukça nadir görülen genetik olarak belirlenmiş bir hastalıktır. Patolojinin gelişim mekanizması henüz tam olarak araştırılmamıştır, ancak sorunun ciddiyeti bir dizi etkili teşhis tekniğinin geliştirilmesine yol açmıştır. Birçok ülkede zorunlu olan yenidoğan taraması, hastalığın zamanında tespit edilip harekete geçilmesini mümkün kılıyor. Genetik olarak belirlenmiş hastalıkların tedavisi bugüne kadar tam olarak gelişmemiş olsa da, terapi hastalığın şiddetli belirtilerinden ve ilerlemesinden kurtulmaya yardımcı olur. Bu durumda ebeveynlerin görevi, patolojiyi mümkün olduğu kadar erken teşhis etmek ve doktorun talimatlarını takip ederek çocuğun konjenital anomaliyle başa çıkmasına yardımcı olmaktır.
Galaktozemi nedir - videoyu izleyin:
