
Cos'è la galattosemia e perché si manifesta in un bambino? Sintomi, diagnosi della malattia. Trattamento della galattosemia nei neonati e misure preventive disponibili.
Il contenuto dell'articolo:- Cos'è la galattosemia
- Sintomi dello sviluppo
- Diagnostica
- Come trattare la galattosemia
- Prevenzione
La galattosemia è un'anomalia ereditaria rara ma ancora comune associata al metabolismo. L'enzimopatia è associata alla disfunzione del metabolismo dei carboidrati, a seguito della quale il corpo accumula galattosio e suoi derivati. Quantità eccessive di galattosio portano allo sviluppo di un quadro clinico. In caso di diagnosi tardiva della galattosemia, i sintomi diventano pronunciati e la prognosi per la normalizzazione delle condizioni del paziente è desolante. Le coppie con predisposizione alla malattia devono conoscere i tipi di galattosemia, i metodi di diagnosi, trattamento e prevenzione e pensare anche alla pianificazione familiare. Le misure tempestive adottate riducono significativamente il rischio di complicanze nel neonato.
Cos'è la galattosemia?
Prima di parlare di raccomandazioni per la galattosemia, è necessario comprendere il ruolo del galattosio nel metabolismo dei carboidrati. Questo zucchero semplice entra nel corpo umano come parte del lattosio, un disaccaride il cui secondo componente è il glucosio. Normalmente, il monosaccaride viene utilizzato come risorsa “energetica” per cellule e composti complessi, materiali “da costruzione” del tessuto nervoso, delle membrane e delle terminazioni nervose.
In una persona sana, il lattosio viene scomposto nei suoi componenti nel tratto digestivo e gradualmente elaborato. Pertanto, nella prima fase del trattamento, il galattosio 1-fosfato viene ottenuto sotto l'azione di uno speciale enzima galattochinasi e ATP. Nella fase successiva, come risultato dello scambio sotto l'azione degli enzimi, si ottengono glucosio 1-fosfato e uridina fosfato galattosio. I monosaccaridi vengono assorbiti molto più facilmente dall'intestino durante la digestione, il che è molto importante per i neonati, poiché il latte è la base della loro dieta.
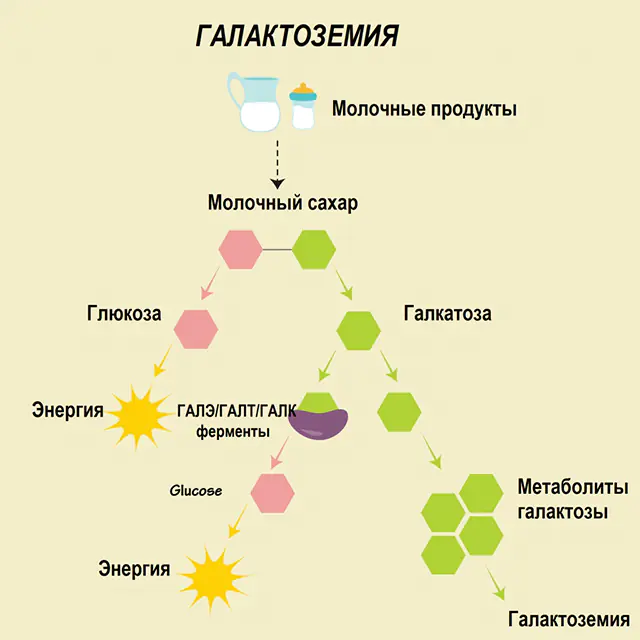
Tre enzimi sono coinvolti nei processi di lavorazione: GALA (galattochinasi), GALT (galattosio 1-fosfato uridiil transferasi), GALE (epimerasi). La produzione carente di almeno uno di questi enzimi nella prima o nella seconda fase del metabolismo dei carboidrati porta all'accumulo di metaboliti, all'intossicazione del corpo e alla successiva manifestazione dei sintomi della galattosemia.
A seconda dell'enzima difettoso, si distinguono i tipi corrispondenti di galattosemia:
- Violazione del GALT- il primo tipo si verifica in un caso ogni 40mila nati; l'analisi per la galattosemia mostrerà una ridotta attività enzimatica;
- Patologia GALC- il secondo tipo si manifesta in un bambino su 500mila, mentre l'attività dell'enzima basico negli eritrociti rientra nel range normale, ma i sintomi della malattia sono pronunciati;
- BURRASCA— la terza tipologia è quella meno registrata (un caso su un milione di persone) ed è caratterizzata da sintomi lievi.
Il tipo più comune di malattia (GALT) si divide in forme, a seconda della genetica della galattosemia:
- Il fallimento è caratteristico dello stadio di trasformazione dell'ammide dell'acido aspartico in aspartato - la patologia è anche chiamata segno di Duarte (Duarte). È interessante notare che una persona con una tale anomalia si sentirà assolutamente sana, sebbene l'attività dell'enzima GALT sia ridotta del 25%-50%.
- Il fallimento della transizione glutammina-arginina si registra più spesso nei caucasici, l'attività enzimatica è solo il 10% della norma, il che provoca un decorso grave della malattia.
- Quando l'enzima è completamente inattivo si verifica un'interruzione della transizione arginina-triptofano, una forma estremamente grave della malattia.
- L'interruzione del processo di trasformazione lisina-asparagina si verifica abbastanza spesso.
- La mancata fermentazione della serina-leucina è stata registrata solo nei rappresentanti della razza negroide ed è associata ad un'attività enzimatica insufficiente nel fegato (a un livello non superiore al 10% della norma generalmente accettata).
L'enzima GALT è presente nell'organismo in quantità eccessive, quindi una leggera diminuzione della sua attività non sempre si manifesta come sintomi clinici. Solo quando l’attività del GALT diminuisce del 50% o più compaiono i primi segni della malattia.
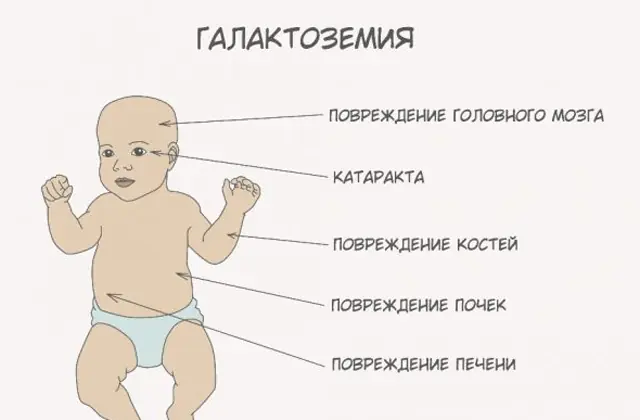
L'intossicazione del corpo con galattosio e metaboliti è pericolosa per lo sviluppo di complicanze a lungo termine: compromissione della funzione motoria, della parola, delle funzioni riproduttive, soprattutto nelle ragazze (nonostante la frequenza delle malattie nei ragazzi e nelle ragazze sia la stessa), ritardo della crescita e altre disfunzioni dello sviluppo.
Nota! La patogenesi della malattia non è stata ancora completamente studiata. Tuttavia, è già stato stabilito che l'accumulo di galattosio nell'organismo è pericoloso non solo a causa del suo dosaggio tossico, ma anche a causa del suo effetto inibitorio su altri enzimi. Senza un trattamento e un controllo adeguati, il paziente può sviluppare la sindrome ipoglicemica.Come già notato, la patologia è un'anomalia congenita. Il tipo di eredità della galattosemia è autosomico recessivo, cioè il bambino riceve geni difettosi dai genitori. Il gene mutato può essere trovato in diversi alleli (forme della condizione): difettoso e normale, quando il difetto è stato trasmesso da un solo genitore. In questo caso, la malattia si manifesta in misura minore. Ma se gli alleli difettosi sono stati trasmessi sia dal padre che dalla madre secondo il tipo di eredità della galattosemia, i primi segni della malattia appariranno entro pochi giorni dalla nascita del bambino. È interessante notare che se una malattia viene diagnosticata in un bambino in una famiglia, la probabilità che la patologia si manifesti nel figlio successivo degli stessi genitori aumenta del 25%.
Una produzione insufficiente di enzimi porta all'accumulo di galattosio nel corpo e, di conseguenza, compaiono segni di galattosemia e le funzioni del sistema escretore vengono interrotte. Senza trattamento entro pochi mesi di vita, una persona muore per insufficienza epatica o infezioni che colpiscono il corpo.
È importante che le principali cause della galattosemia (componente ereditaria) possano essere stabilite anche prima della nascita del bambino, ma i medici non escludono l'influenza di altri indicatori sullo sviluppo della patologia:
- altre mutazioni genetiche;
- tessuto interessato del fegato e del sistema nervoso centrale;
- accumulo di liquido nel cristallino dell'occhio, visione offuscata.
Eppure, se a un paziente viene diagnosticata la galattosemia, la genetica gioca un ruolo chiave. I fattori concomitanti sono importanti solo nei pazienti eterozigoti, cioè quelli in cui il gene difettoso è stato trasmesso da un solo genitore. Nei pazienti omozigoti, la patologia stessa è grave e richiede un trattamento altamente specializzato.
Sintomi dello sviluppo della galattosemia
In base alla gravità delle manifestazioni dei segni di galattosemia, si distinguono tre gradi della malattia:
- leggero- solitamente rilevato accidentalmente a causa di un'intolleranza al latte;
- media- i primi segni compaiono solo dopo aver bevuto il latte;
- pesante— i sintomi della patologia si sovrappongono ad altri malfunzionamenti del corpo (accumulo di liquidi nella cavità addominale, sepsi).
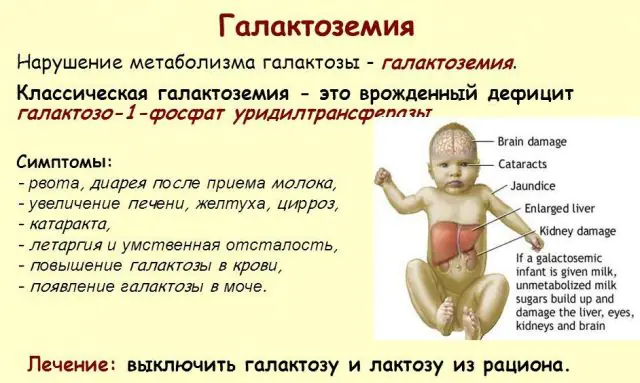
La patologia GALT classica si manifesta nella sua forma più grave. I primi segni della malattia diventeranno visibili dopo la nascita del bambino e la sua prima poppata, poiché il latte costituisce la dieta principale del neonato. Dopo aver mangiato, avverte vomito, frequenti movimenti intestinali e una maggiore sonnolenza sullo sfondo di letargia costante e ipotensione muscolare. Nonostante l'allattamento al seno regolare, non si verifica alcun aumento di peso. I disturbi digestivi sono uno dei primi segni di disfunzione.
Quando il corpo viene intossicato, i segni di danno epatico diventano pronunciati: ittero, ingrossamento del fegato. Nel giro di poche settimane, il bambino sviluppa la cataratta e dopo alcuni mesi, a causa dell'intossicazione dei tessuti nervosi, si notano disturbi nelle funzioni psicomotorie. L'attività dei tubuli renali viene interrotta, a seguito della quale nelle urine appare una riduzione dello zucchero e si nota una diminuzione della coagulazione del sangue.
Senza la terapia necessaria, i sintomi peggiorano man mano che il corpo del bambino viene intossicato e aumenta il rischio per il normale sviluppo e persino per la vita umana. Il 20-30% dei bambini, se il trattamento viene rifiutato o la malattia viene diagnosticata tardivamente, muore di sepsi, che si sviluppa a causa dell'attività leucocitotica. Coloro che sopravvivono soffrono di insufficienza renale e problemi di sviluppo psicomotorio.
In molti modi, il grado di manifestazione della patologia dipende dalla causa ereditaria. La galattosemia si manifesta quando si verifica il deficit di GALT. Pertanto, il segno di Duhart all'inizio può rivelarsi solo come ittero prolungato (fino a 2 mesi dopo la nascita), la cataratta e la disfunzione epatica vengono diagnosticate un po' più tardi. Ma se l'attività dell'enzima GALT è almeno del 50%, i sintomi clinici potrebbero non comparire.
La mancanza dell'enzima GALA non è così pronunciata. Il sintomo principale della malattia è la cataratta progressiva e nel tempo possono comparire anche monosaccaridi nelle urine. Allo stesso tempo, la perdita di peso del bambino è insignificante e le capacità psicomotorie si sviluppano entro limiti normali.
La carenza di GALE è inizialmente estremamente rara, ma i sintomi si dividono ancora in due forme: benigna, quando la carenza dell'enzima colpisce le cellule del sangue, e generalizzata, quando la carenza colpisce tutti i tessuti del corpo. Nel primo caso, solo l'analisi della galattosemia aiuterà a individuare la malattia nelle fasi iniziali; solo con il tempo si noteranno disfunzioni delle capacità psicomotorie e dello sviluppo del linguaggio. La seconda forma è simile nei sintomi al deficit di GALT, ma in questo caso è presente anche un ingrossamento della milza. Anche con il trattamento di una forma grave di malattia GALE, col tempo il bambino sperimenta un ritardo nello sviluppo psicomotorio, sordità e problemi alla vista.
Diagnosi di galattosemia
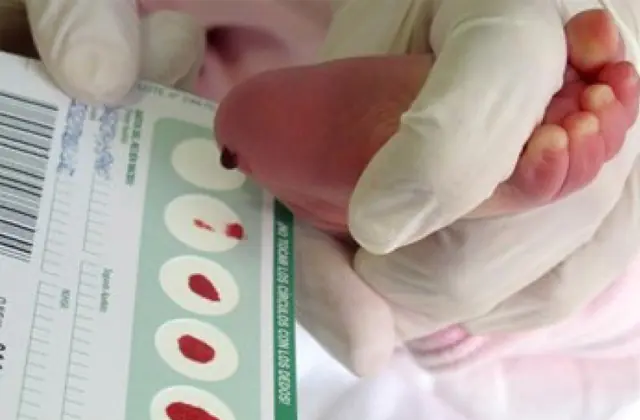
Lo screening neonatale, introdotto in numerosi paesi, consente di identificare fino a 50 malattie congenite nelle fasi iniziali, inclusa la produzione insufficiente di enzimi attivamente coinvolti nel metabolismo. Se per qualsiasi motivo lo screening non viene effettuato, la malattia viene identificata sulla base delle caratteristiche aggregate.
Pertanto, se si sospetta la galattosemia, le raccomandazioni del medico vengono formulate solo dopo una diagnosi dettagliata. Un piccolo paziente deve sottoporsi a:
- esame generale: se sono presenti segni di ittero;
- vengono valutati i segni generali: la quantità e la qualità dei movimenti intestinali, la presenza o l'assenza di ipotonicità muscolare, problemi con le capacità psicomotorie;
- esami di laboratorio: esame del sangue per biochimica, esame del sangue generale, esame delle urine per zucchero e proteine;
- ulteriore esame delle condizioni del fegato e dei reni.
La diagnostica del DNA è obbligatoria per rilevare le mutazioni GALT. Se il bambino presenta sintomi della malattia e l'attività degli enzimi GALT secondo i risultati del test è normale, viene eseguita la diagnosi del DNA per l'enzima GALE.
I medici possono prescrivere una serie di test aggiuntivi per differenziare la diagnosi. È necessaria una diagnosi esclusiva per confermare o smentire la presenza di diabete mellito e altre patologie associate ad alti livelli di zucchero, nonché malattie che causano l'ingrossamento del fegato.
Come trattare la galattosemia?

Una volta che lo screening neonatale e altri esami hanno confermato la presenza di galattosemia, viene prescritto il trattamento. È necessario comprendere che poiché la malattia è causata da una predisposizione genetica, una cura completa in questa fase dello sviluppo medico è impossibile. La terapia ha lo scopo di ridurre le manifestazioni cliniche della patologia, minimizzando la progressione della malattia e lo sviluppo di complicanze.
Il principale metodo di trattamento è una corretta alimentazione per la galattosemia. Tutti i prodotti contenenti latte, anche pane, dolci, salsicce e altri, sono completamente esclusi dalla dieta del paziente. Vengono eliminati anche i prodotti di origine vegetale e animale contenenti oligosaccaridi: legumi, soia, spinaci, cioccolato, frutta secca, fegato, cervello, uova e derivati di questi prodotti. Una rigorosa terapia dietetica è l'unico modo per prevenire l'intossicazione del corpo.
Per i neonati è specializzata anche la nutrizione per la galattosemia. Il latte materno viene gradualmente eliminato completamente dalla dieta e sostituito con formule senza lattosio e formulazioni a base di proteine isolate della soia. Il volume degli ingredienti nutrizionali in questo caso dovrebbe corrispondere agli standard per un bambino sano. Se un bambino presenta sintomi di allergia alle proteine isolate della soia, vengono selezionate miscele a base di idrolizzati di caseina. La prima assunzione di miscela alimentare dovrebbe corrispondere a 1/10 del fabbisogno nutrizionale giornaliero del bambino. Entro 5-7 giorni il latte materno viene completamente eliminato dalla dieta.
I primi alimenti complementari per i bambini con patologia vengono prescritti a partire dai 4 mesi. La dieta viene ampliata con succhi di frutta (mela, pera e altri). Dopo mezzo mese vengono introdotte le puree di frutta. Fino a 1 anno, il volume di succo e purea consumato al giorno aumenta a 30-50 ml. La prima purea di verdure in acqua (esclusi i legumi) viene introdotta a 5 mesi. Da 5,5 mesi, a un bambino possono essere somministrati cereali commerciali senza latticini (grano saraceno, riso, mais). Il bambino può provare prodotti a base di carne dall'età di 6 mesi, la preferenza dovrebbe essere data a cibi in scatola specializzati che non contengono latte o prodotti contenenti latte.
Quando si scelgono prodotti alimentari specializzati per neonati, è necessario leggere attentamente le etichette. I prodotti che contengono non più di 5 mg di galattosio per 100 g sono considerati sicuri per un bambino.Se la quantità di galattosio supera i 20 mg, è severamente vietato somministrare il prodotto a un bambino.
L'adeguatezza del trattamento per la galattosemia viene verificata mediante un esame di controllo una volta ogni trimestre. Viene anche testata l'efficacia del trattamento dei sintomi associati alla patologia.
Importante! Particolare attenzione è rivolta alla selezione dei farmaci per tali pazienti. Alcuni farmaci contengono lattosio nella loro composizione, mentre altri possono inibire la rimozione dei prodotti metabolici dal fegato, il che generalmente aggrava i sintomi dell'anomalia congenita ed è inaccettabile nell'ambito di un trattamento competente.Prevenzione della galattosemia

Misure preventive specializzate per la malattia non sono ancora state sviluppate. Se si riscontra che i genitori hanno una predisposizione alla patologia, si consiglia di pianificare il concepimento con ulteriori consultazioni con un genetista. Durante la gravidanza, dovresti anche sottoporti a una serie di esami (biopsia dei villi coriali a 10-12 settimane e test del liquido amniotico a 15-18 settimane) per rilevare mutazioni GALT e GALE nei geni.
Nonostante lo sviluppo di un trattamento sintomatico abbastanza efficace della patologia, le prospettive a lungo termine per lo sviluppo del bambino sono difficili da prevedere. Pertanto, gli indicatori di sviluppo fisico del paziente saranno inferiori a quelli dei coetanei, è probabile che si sviluppino disturbi della parola e della coordinazione, una maggiore fragilità ossea e, nelle ragazze, disfunzione ovarica.
Dall'età di 5 anni, a un bambino può essere consigliato di assumere vitamine e farmaci contenenti ATP per prevenire la progressione della malattia. Dall'età di 12 anni, alle ragazze viene prescritta una terapia ormonale per compensare la disfunzione ovarica.
La galattosemia è una malattia geneticamente determinata abbastanza rara. Il meccanismo di sviluppo della patologia non è stato ancora studiato a fondo, ma la gravità del problema ha portato allo sviluppo di una serie di tecniche diagnostiche efficaci. Lo screening neonatale, obbligatorio in numerosi paesi, consente di individuare tempestivamente la malattia e di intervenire. E sebbene il trattamento delle malattie geneticamente determinate fino ad oggi non sia stato completamente sviluppato, la terapia aiuta a eliminare i sintomi gravi della malattia e il suo progresso. Il compito dei genitori in questo caso è diagnosticare la patologia il prima possibile e seguire le indicazioni del medico, aiutando così il bambino ad affrontare l’anomalia congenita.
Cos'è la galattosemia: guarda il video:
