
Hva er galaktosemi og hvorfor oppstår det hos et barn? Symptomer, diagnose av sykdommen. Behandling av galaktosemi hos nyfødte og tilgjengelige forebyggende tiltak.
Innholdet i artikkelen:- Hva er galaktosemi
- Utviklingssymptomer
- Diagnostikk
- Hvordan behandle galaktosemi
- Forebygging
Galaktosemi er en sjelden, men fortsatt vanlig arvelig abnormitet assosiert med metabolisme. Enzymopati er assosiert med dysfunksjon av karbohydratmetabolismen, som et resultat av at kroppen akkumulerer galaktose og dets derivater. For store mengder galaktose fører til utvikling av et klinisk bilde. Ved sen diagnose av galaktosemi blir symptomene uttalte, og prognosen for normalisering av pasientens tilstand er dyster. Par med en disposisjon for sykdommen trenger å vite om typer galaktosemi, metoder for diagnose, behandling og forebygging, og også tenke på familieplanlegging. Rettidige tiltak som er tatt, reduserer risikoen for komplikasjoner hos den nyfødte betydelig.
Hva er galaktosemi?
Før du snakker om anbefalinger for galaktosemi, er det nødvendig å forstå rollen til galaktose i karbohydratmetabolismen. Dette enkle sukkeret kommer inn i menneskekroppen som en del av laktose, et disakkarid hvor den andre komponenten er glukose. Normalt brukes monosakkaridet som "energi" ressurser for celler og komplekse forbindelser, "bygge" materialer av nervevev, membraner og nerveender.
Hos en sunn person brytes laktose ned til komponentene i fordøyelseskanalen og behandles gradvis. Således, i det første trinnet av behandlingen, oppnås galaktose 1-fosfat under påvirkning av et spesielt enzym galaktokinase og ATP. På neste trinn, som et resultat av utveksling under påvirkning av enzymer, oppnås glukose 1-fosfat og uridinfosfatgalaktose. Monosakkarider absorberes mye lettere av tarmene under fordøyelsen, noe som er veldig viktig for nyfødte, siden melk er grunnlaget for kostholdet deres.
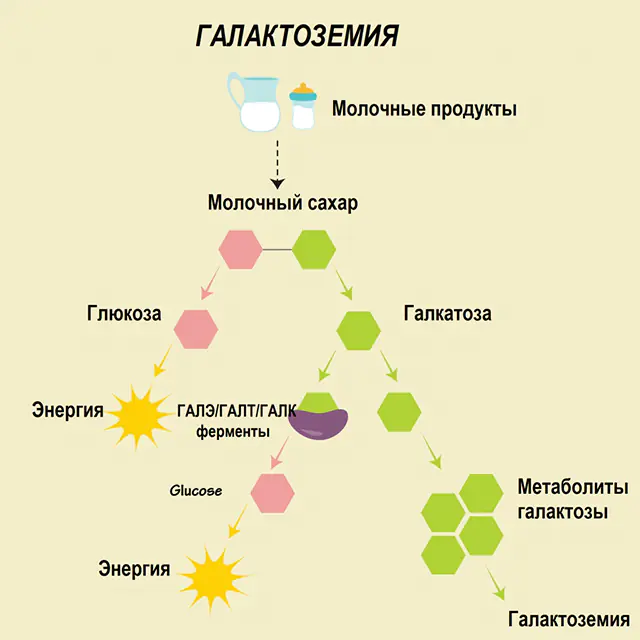
Tre enzymer er involvert i prosesseringsprosessene - GALA (galaktokinase), GALT (galaktose 1-fosfat uridiyl transferase), GALE (epimerase). Mangelfull produksjon av minst ett av disse enzymene i det første eller andre stadiet av karbohydratmetabolismen fører til akkumulering av metabolitter, forgiftning av kroppen og den påfølgende manifestasjonen av symptomer på galaktosemi.
Avhengig av det defekte enzymet skilles de tilsvarende typene galaktosemi ut:
- GALT-brudd- den første typen forekommer i ett tilfelle per 40 tusen nyfødte; analyse for galaktosemi vil vise redusert enzymaktivitet;
- GALC-patologi- den andre typen forekommer hos ett barn av 500 tusen, mens aktiviteten til det grunnleggende enzymet i erytrocytter er innenfor normalområdet, men symptomene på sykdommen er uttalt;
- GALE— den tredje typen ble registrert minst (ett tilfelle per million mennesker) og er preget av milde symptomer.
Den vanligste typen sykdom (GALT) er delt inn i former, avhengig av genetikken til galaktosemi:
- Svikten er karakteristisk for stadiet av transformasjon av asparaginsyreamid til aspartat - patologien kalles også Duartes tegn (Duarte). Det er bemerkelsesverdig at en person med en slik anomali vil føle seg helt sunn, selv om aktiviteten til GALT-enzymet reduseres med 25% -50%.
- Svikt i glutamin-arginin-overgangen er oftest registrert hos kaukasiere; enzymaktivitet er bare 10% av normen, noe som provoserer et alvorlig sykdomsforløp.
- En forstyrrelse av arginin-tryptofan-overgangen oppstår når enzymet er fullstendig inaktivt, en ekstremt alvorlig form av sykdommen.
- Forstyrrelse av lysin-asparagin-transformasjonsprosessen forekommer ganske ofte.
- Svikt i serin-leucin-gjæring ble registrert bare hos representanter for den negroide rasen og er assosiert med utilstrekkelig enzymaktivitet i leveren (på et nivå på ikke mer enn 10% av den generelt aksepterte normen).
GALT-enzymet er tilstede i kroppen i store mengder, så en liten reduksjon i aktiviteten viser seg ikke alltid som kliniske symptomer. Først når GALT-aktiviteten synker med 50 prosent eller mer vil de første tegn på sykdommen vises.
Forgiftning av kroppen med galaktose og metabolitter er farlig for utviklingen av komplikasjoner på lang sikt: nedsatt motorisk funksjon, tale, reproduktive funksjoner, spesielt hos jenter (til tross for at frekvensen av sykdommer hos gutter og jenter er den samme), veksthemming og andre utviklingssvikt.
Merk! Patogenesen til sykdommen er ennå ikke fullt ut studert. Imidlertid har det allerede blitt fastslått at akkumulering av galaktose i kroppen er farlig, ikke bare på grunn av dens giftige dosering, men også på grunn av dens hemmende effekt på andre enzymer. Uten riktig behandling og kontroll kan pasienten utvikle hypoglykemisk syndrom.Som allerede nevnt, er patologien en medfødt anomali. Arten av arv av galaktosemi er autosomal recessiv, det vil si at barnet mottar defekte gener fra foreldrene. Det muterte genet kan finnes i forskjellige alleler (former av tilstanden) - defekte og normale, når defekten ble overført fra bare en forelder. I dette tilfellet manifesterer sykdommen seg i mindre grad. Men hvis defekte alleler har blitt overført fra både far og mor i samsvar med arvetypen for galaktosemi, vil de første tegnene på sykdommen vises innen få dager etter fødselen av babyen. Det er bemerkelsesverdig at hvis en sykdom blir diagnostisert hos ett barn i en familie, øker sannsynligheten for at patologien oppstår hos det neste barnet fra de samme foreldrene med 25%.
Utilstrekkelig produksjon av enzymer fører til akkumulering av galaktose i kroppen, og som et resultat vises tegn på galaktosemi og funksjonene til utskillelsessystemet blir forstyrret. Uten behandling innen noen få måneder av livet, dør en person av leversvikt eller infeksjoner som påvirker kroppen.
Det er viktig at hovedårsakene til galaktosemi (arvelig komponent) kan fastslås allerede før fødselen av barnet, men leger utelukker ikke påvirkningen fra andre indikatorer på utviklingen av patologien:
- andre genmutasjoner;
- påvirket vev i leveren og sentralnervesystemet;
- opphopning av væske i øyelinsen, tåkesyn.
Og likevel, hvis en pasient blir diagnostisert med galaktosemi, spiller genetikk en nøkkelrolle. Samtidige faktorer er bare viktige hos heterozygote pasienter, det vil si de der det defekte genet ble overført fra bare en forelder. Hos homozygote pasienter er selve patologien alvorlig og krever høyt spesialisert behandling.
Symptomer på utvikling av galaktosemi
I henhold til alvorlighetsgraden av manifestasjonene av tegn på galaktosemi, skilles tre grader av sykdommen:
- lys- vanligvis oppdaget ved et uhell på grunn av melkintoleranse;
- gjennomsnitt- de første tegnene vises først etter å ha drukket melk;
- tung— symptomene på patologien er lagt over andre funksjonsfeil i kroppen (væskeansamling i bukhulen, sepsis).
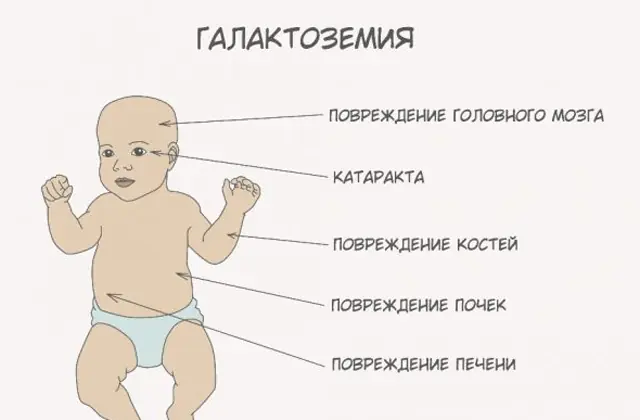
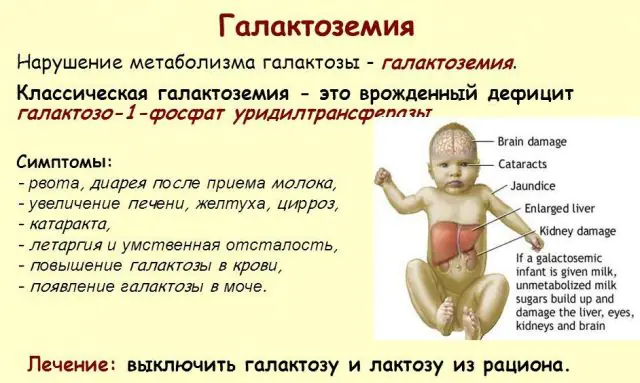
Klassisk GALT-patologi manifesterer seg i sin mest alvorlige form. De første tegnene på sykdommen vil bli synlige etter fødselen av barnet og hans første fôring, siden melk utgjør hoveddietten til den nyfødte. Etter fôring opplever han oppkast, hyppige avføringer og økt døsighet på bakgrunn av konstant sløvhet og muskelhypotensjon. Til tross for vanlig amming er det ingen vektøkning. Fordøyelsessykdommer er et av de første tegnene på funksjonssvikt.
Når kroppen blir beruset, blir tegn på leverskade uttalt - gulsott, forstørrelse av leveren. I løpet av noen få uker utvikler barnet grå stær, og etter noen måneder, på grunn av forgiftning av nervevevet, noteres forstyrrelser i psykomotoriske funksjoner. Aktiviteten til nyretubuli blir forstyrret, som et resultat av at reduserende sukker vises i urinen, og redusert blodpropp noteres.
Uten nødvendig terapi forverres symptomene når babyens kropp blir beruset, og risikoen for normal utvikling og til og med menneskeliv øker. 20-30 % av barna dør, hvis behandling nektes eller sykdommen diagnostiseres sent, av sepsis, som utvikler seg på grunn av leukocytotisk aktivitet. De som overlever lider av nyresvikt og psykomotoriske utviklingsproblemer.
På mange måter avhenger graden av manifestasjon av patologien av den arvelige årsaken. Galaktosemi manifesterer seg når GALT-mangel oppstår. Dermed kan Duharts tegn først avsløre seg som langvarig gulsott (opptil 2 måneder etter fødselen), grå stær og leverdysfunksjon diagnostiseres litt senere. Men hvis aktiviteten til GALT-enzymet er minst 50 %, kan det hende at kliniske symptomer ikke vises.
Mangelen på GALA-enzym er ikke så uttalt. Hovedsymptomet på sykdommen er progressiv grå stær, og monosakkarider kan også oppstå i urinen over tid. Samtidig er babyens vekttap ubetydelig, og psykomotoriske ferdigheter utvikler seg innenfor normalområdet.
GALE-mangel er i utgangspunktet ekstremt sjelden, men symptomene er fortsatt delt inn i to former: godartet, når mangel på enzymet påvirker blodceller, og generalisert, når mangelen påvirker alle vev i kroppen. I det første tilfellet vil bare en analyse for galaktosemi bidra til å oppdage sykdommen i de tidlige stadiene; bare med tiden vil dysfunksjon av psykomotoriske ferdigheter og taleutvikling bli notert. Den andre formen ligner i sine symptomer på GALT-mangel, men i dette tilfellet er det også en forstørrelse av milten. Selv med behandling av en alvorlig form for GALE-sykdom, opplever barnet over tid forsinket psykomotorisk utvikling, døvhet og synsproblemer.
Diagnose av galaktosemi
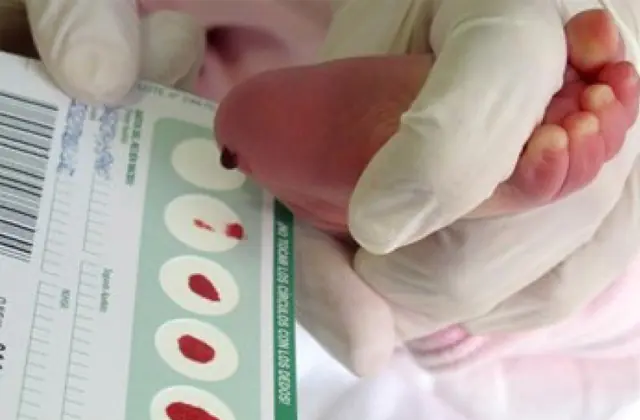
Neonatal screening, introdusert i en rekke land, gjør det mulig å identifisere opptil 50 medfødte sykdommer i tidlige stadier, inkludert utilstrekkelig produksjon av enzymer som er aktivt involvert i metabolismen. Hvis screening av en eller annen grunn ikke utføres, identifiseres sykdommen basert på aggregerte egenskaper.
Så hvis det er mistanke om galaktosemi, gis legens anbefalinger først etter en detaljert diagnose. En liten pasient må gjennomgå:
- generell undersøkelse - om tegn på gulsott er tilstede;
- generelle tegn vurderes - kvantiteten og kvaliteten på avføring, tilstedeværelse eller fravær av muskelhypotoni, problemer med psykomotoriske ferdigheter;
- laboratorietester - blodprøve for biokjemi, generell blodprøve, urinprøve for sukker og protein;
- tilleggsundersøkelse av tilstanden til leveren og nyrene.
DNA-diagnostikk er obligatorisk for å oppdage GALT-mutasjoner. Dersom barnet viser symptomer på sykdommen, og aktiviteten til GALT-enzymer ifølge testresultater er normal, utføres DNA-diagnostikk for GALE-enzymet.
Leger kan foreskrive en rekke tilleggstester for å skille diagnosen. Eksklusiv diagnose er nødvendig for å bekrefte eller tilbakevise tilstedeværelsen av diabetes mellitus og andre patologier forbundet med høye sukkernivåer, samt sykdommer som forårsaker leverforstørrelse.
Hvordan behandle galaktosemi?

Når neonatal screening og andre undersøkelser har bekreftet tilstedeværelsen av galaktosemi, foreskrives behandling. Det er nødvendig å forstå at siden sykdommen er forårsaket av en genetisk predisposisjon, er en fullstendig kur på dette stadiet av medisinsk utvikling umulig. Terapi er rettet mot å redusere de kliniske manifestasjonene av patologien, minimere utviklingen av sykdommen og utviklingen av komplikasjoner.
Hovedbehandlingsmetoden er riktig ernæring for galaktosemi. Alle melkeholdige produkter, til og med brød, søtsaker, pølser og andre, er fullstendig ekskludert fra pasientens kosthold. Produkter av plante- og animalsk opprinnelse som inneholder oligosakkarider fjernes også: belgfrukter, soya, spinat, sjokolade, nøtter, lever, hjerner, egg og derivater av disse produktene. Strenge kostholdsterapi er den eneste måten å forhindre forgiftning av kroppen.
For nyfødte er ernæring for galaktosemi også spesialisert. Morsmelk elimineres gradvis fullstendig fra kostholdet og erstattes med laktosefrie formuleringer og formuleringer basert på soyaproteinisolat. Volumet av næringsingredienser i dette tilfellet bør samsvare med standardene for et sunt barn. Hvis en baby viser symptomer på allergi mot soyaproteinisolat, velges blandinger basert på kaseinhydrolysater. Det første inntaket av matblanding bør være 1/10 av barnets daglige ernæringsbehov. I løpet av 5-7 dager er morsmelk fullstendig eliminert fra kostholdet.
De første komplementære matvarene for barn med patologi er foreskrevet fra 4 måneder. Kostholdet utvides med fruktjuicer (eple, pære og andre). Etter en halv måned introduseres fruktpuréer. Opptil 1 år øker volumet av juice og puré per dag til 30-50 ml. Den første grønnsakspuréen i vann (unntatt belgfrukter) introduseres ved 5 måneder. Fra 5,5 måneder kan et barn gis melkefrie kommersielle frokostblandinger (bokhvete, ris, mais). Babyen kan prøve kjøttprodukter fra 6 måneders alder; spesialisert hermetikk som ikke inneholder melk eller melkeholdige produkter bør foretrekkes.
Når du velger spesialiserte matvarer for spedbarn, må du lese etikettene nøye. Produkter som ikke inneholder mer enn 5 mg galaktose per 100 g anses som trygge for en baby. Hvis det er mer enn 20 mg galaktose, er det strengt forbudt å gi produktet til et barn.
Tilstrekkelig behandling for galaktosemi kontrolleres ved en kontrollundersøkelse en gang i kvartalet. Effektiviteten av behandlingen av medfølgende symptomer på patologi blir også testet.
Viktig! Spesiell oppmerksomhet rettes mot valg av medisiner for slike pasienter. Noen legemidler inneholder laktose i sammensetningen, mens andre kan hemme fjerning av metabolske produkter fra leveren, noe som generelt forverrer symptomene på den medfødte anomalien og er uakseptabel innenfor rammen av kompetent behandling.Forebygging av galaktosemi

Spesialiserte forebyggende tiltak for sykdommen er ennå ikke utviklet. Hvis det er funnet at foreldre har en disposisjon for patologien, anbefales det å planlegge unnfangelse med ytterligere konsultasjoner med en genetiker. Under graviditet bør du også gjennomgå en rekke undersøkelser (chorionic villus biopsi ved 10-12 uker og fostervannsprøve ved 15-18 uker) for å oppdage GALT- og GALE-mutasjoner i gener.
Til tross for den utviklet ganske effektive symptomatisk behandling av patologien, er langsiktige utsikter for barnets utvikling vanskelig å forutsi. Dermed vil pasientens fysiske utviklingsindikatorer være lavere enn jevnaldrende, tale- og koordinasjonsforstyrrelser vil sannsynligvis utvikle seg, økt benskjørhet, og hos jenter - eggstokkdysfunksjon.
Fra 5-årsalderen kan et barn bli anbefalt å ta vitaminer og ATP-holdige legemidler for å forhindre utviklingen av sykdommen. Fra de er 12 år blir jenter foreskrevet hormonbehandling for å kompensere for eggstokkdysfunksjon.
Galaktosemi er en ganske sjelden genetisk betinget sykdom. Mekanismen for utvikling av patologien har ennå ikke blitt grundig studert, men alvorlighetsgraden av problemet har ført til utviklingen av en rekke effektive diagnostiske teknikker. Neonatal screening, obligatorisk i en rekke land, gjør det mulig å oppdage sykdommen i tide og iverksette tiltak. Og selv om behandlingen av genetisk betingede sykdommer ikke er fullt utviklet til dags dato, hjelper terapi med å bli kvitt alvorlige symptomer på sykdommen og dens fremgang. Foreldrenes oppgave i dette tilfellet er å diagnostisere patologien så tidlig som mulig og følge legens instruksjoner, og dermed hjelpe barnet med å takle den medfødte anomalien.
Hva er galaktosemi - se videoen:
