
Czym jest galaktozemia i dlaczego występuje u dziecka? Objawy, rozpoznanie choroby. Leczenie galaktozemii u noworodków i dostępne środki zapobiegawcze.
Treść artykułu:- Co to jest galaktozemia
- Objawy rozwojowe
- Diagnostyka
- Jak leczyć galaktozemię
- Zapobieganie
Galaktozemia jest rzadką, ale wciąż częstą dziedziczną chorobą związaną z metabolizmem. Enzymopatia wiąże się z zaburzeniem metabolizmu węglowodanów, w wyniku czego w organizmie gromadzi się galaktoza i jej pochodne. Nadmierne ilości galaktozy prowadzą do powstania obrazu klinicznego. W przypadku późnego rozpoznania galaktozemii objawy stają się wyraźne, a rokowania dotyczące normalizacji stanu pacjenta są ponure. Pary z predyspozycją do tej choroby muszą wiedzieć o rodzajach galaktozemii, sposobach diagnozowania, leczenia i profilaktyki, a także myśleć o planowaniu rodziny. Podjęte w odpowiednim czasie działania znacznie zmniejszają ryzyko powikłań u noworodka.
Co to jest galaktozemia?
Zanim zaczniemy mówić o zaleceniach dotyczących galaktozemii, należy zrozumieć rolę galaktozy w metabolizmie węglowodanów. Ten cukier prosty dostaje się do organizmu ludzkiego w ramach laktozy, disacharydu, którego drugim składnikiem jest glukoza. Zwykle monosacharyd jest wykorzystywany jako źródło „energii” dla komórek i związków złożonych, materiałów „budowlanych” tkanki nerwowej, błon i zakończeń nerwowych.
U zdrowego człowieka laktoza rozkładana jest w przewodzie pokarmowym na składniki i stopniowo przetwarzana. Zatem w pierwszym etapie przetwarzania pod działaniem specjalnego enzymu galaktokinazy i ATP otrzymuje się galaktozo-1-fosforan. W kolejnym etapie, w wyniku wymiany pod działaniem enzymów, otrzymuje się glukozo-1-fosforan i galaktozę urydynofosforanową. Monosacharydy są znacznie łatwiej wchłaniane w jelitach podczas trawienia, co jest bardzo ważne dla noworodków, ponieważ mleko jest podstawą ich diety.
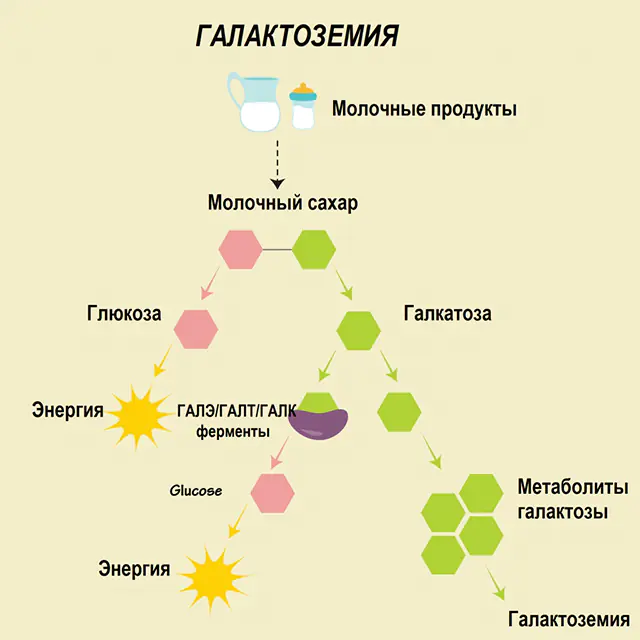
W procesach przetwarzania biorą udział trzy enzymy - GALA (galaktokinaza), GALT (urydiylotransferaza galaktozo-1-fosforanowa), GALE (epimeraza). Niedobór przynajmniej jednego z tych enzymów na pierwszym lub drugim etapie metabolizmu węglowodanów prowadzi do gromadzenia się metabolitów, zatrucia organizmu i późniejszego ujawnienia się objawów galaktozemii.
W zależności od wadliwego enzymu wyróżnia się odpowiednie typy galaktozemii:
- Naruszenie GALT- pierwszy typ występuje w jednym przypadku na 40 tysięcy noworodków, analiza w kierunku galaktozemii wykaże zmniejszoną aktywność enzymu;
- Patologia GALC- drugi typ występuje u jednego dziecka na 500 tysięcy, podczas gdy aktywność podstawowego enzymu w erytrocytach mieści się w granicach normy, ale objawy choroby są wyraźne;
- WICHURA— trzeci typ występował najrzadziej (jeden przypadek na milion osób) i charakteryzował się łagodnymi objawami.
Najczęstszy typ choroby (GALT) dzieli się na formy, w zależności od genetyki galaktozemii:
- Niepowodzenie jest charakterystyczne dla etapu przemiany amidu kwasu asparaginowego w asparaginian – patologia ta nazywana jest także objawem Duarte’a (Duarte). Warto zauważyć, że osoba z taką anomalią będzie czuła się całkowicie zdrowa, chociaż aktywność enzymu GALT jest zmniejszona o 25% -50%.
- Niepowodzenie przejścia glutamina-arginina jest najczęściej odnotowywane u osób rasy kaukaskiej, aktywność enzymatyczna wynosi tylko 10% normy, co powoduje ciężki przebieg choroby.
- Zakłócenie przejścia arginina-tryptofan następuje, gdy enzym jest całkowicie nieaktywny, co jest niezwykle ciężką postacią choroby.
- Zakłócenie procesu przemiany lizyny-asparaginy zdarza się dość często.
- Niewydolność fermentacji serynowo-leucynowej odnotowano jedynie u przedstawicieli rasy Negroidów i wiąże się ona z niedostateczną aktywnością enzymów w wątrobie (na poziomie nie większym niż 10% ogólnie przyjętej normy).
Enzym GALT występuje w organizmie w nadmiernych ilościach, dlatego nieznaczny spadek jego aktywności nie zawsze objawia się objawami klinicznymi. Dopiero gdy aktywność GALT spadnie o 50 procent lub więcej, pojawią się pierwsze oznaki choroby.
Zatrucie organizmu galaktozą i metabolitami jest niebezpieczne w przypadku długotrwałego rozwoju powikłań: upośledzenia funkcji motorycznych, mowy, funkcji rozrodczych, zwłaszcza u dziewcząt (mimo że częstość występowania chorób u chłopców i dziewcząt jest taka sama), opóźnienie wzrostu i inne zaburzenia rozwojowe.
Notatka! Patogeneza choroby nie została jeszcze w pełni poznana. Ustalono jednak już, że gromadzenie się galaktozy w organizmie jest niebezpieczne nie tylko ze względu na toksyczne dawkowanie, ale także ze względu na hamujący wpływ na inne enzymy. Bez odpowiedniego leczenia i kontroli u pacjenta może rozwinąć się zespół hipoglikemiczny.Jak już wspomniano, patologia jest wrodzoną anomalią. Rodzaj dziedziczenia galaktozemii jest autosomalny recesywny, co oznacza, że dziecko otrzymuje wadliwe geny od rodziców. Zmutowany gen można znaleźć w różnych allelach (postaciach choroby) – wadliwych i normalnych, gdy wada została przekazana tylko od jednego rodzica. W tym przypadku choroba objawia się w mniejszym stopniu. Jeśli jednak wadliwe allele zostały przekazane zarówno od ojca, jak i od matki, zgodnie z rodzajem dziedziczenia galaktozemii, pierwsze oznaki choroby pojawią się w ciągu kilku dni po urodzeniu dziecka. Warto zauważyć, że jeśli choroba zostanie zdiagnozowana u jednego dziecka w rodzinie, prawdopodobieństwo wystąpienia patologii u kolejnego dziecka tych samych rodziców wzrasta o 25%.
Niedostateczna produkcja enzymów prowadzi do gromadzenia się galaktozy w organizmie, w efekcie czego pojawiają się objawy galaktozemii i zaburzenia funkcji układu wydalniczego. Bez leczenia w ciągu kilku miesięcy życia osoba umiera z powodu niewydolności wątroby lub infekcji atakujących organizm.
Ważne jest, aby główne przyczyny galaktozemii (składnik dziedziczny) można było ustalić jeszcze przed urodzeniem dziecka, ale lekarze nie wykluczają wpływu innych wskaźników na rozwój patologii:
- inne mutacje genów;
- dotknięta tkanka wątroby i ośrodkowego układu nerwowego;
- nagromadzenie płynu w soczewce oka, niewyraźne widzenie.
A jednak w przypadku zdiagnozowania u pacjenta galaktozemii kluczową rolę odgrywa genetyka. Czynniki współistniejące są ważne tylko u pacjentów heterozygotycznych, to znaczy tych, u których wadliwy gen został przekazany tylko od jednego rodzica. U pacjentów homozygotycznych sama patologia jest ciężka i wymaga wysokospecjalistycznego leczenia.
Objawy rozwoju galaktozemii
W zależności od nasilenia objawów galaktozemii wyróżnia się trzy stopnie choroby:
- światło- zwykle wykrywane przypadkowo w wyniku nietolerancji mleka;
- przeciętny- pierwsze objawy pojawiają się dopiero po wypiciu mleka;
- ciężki— objawy patologii nakładają się na inne zaburzenia w organizmie (nagromadzenie płynu w jamie brzusznej, posocznica).
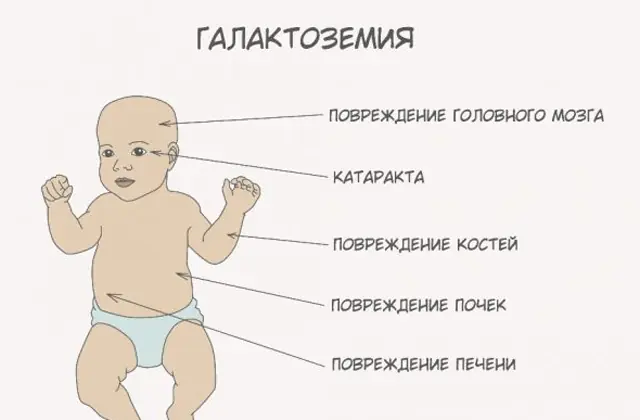
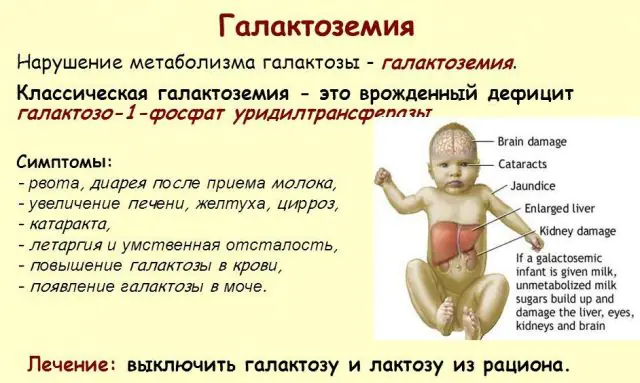
Klasyczna patologia GALT objawia się w najcięższej postaci. Pierwsze oznaki choroby staną się widoczne po urodzeniu dziecka i jego pierwszym karmieniu, ponieważ mleko stanowi główną dietę noworodka. Po karmieniu doświadcza wymiotów, częstych wypróżnień i zwiększonej senności na tle ciągłego letargu i niedociśnienia mięśni. Pomimo regularnego karmienia piersią nie następuje przyrost masy ciała. Zaburzenia trawienia są jednym z pierwszych objawów dysfunkcji.
W miarę odurzenia organizmu pojawiają się oznaki uszkodzenia wątroby - żółtaczka, powiększenie wątroby. W ciągu kilku tygodni u dziecka rozwija się zaćma, a po kilku miesiącach, w wyniku zatrucia tkanek nerwowych, zauważa się zaburzenia funkcji psychomotorycznych. Aktywność kanalików nerkowych zostaje zakłócona, w wyniku czego w moczu pojawia się cukier redukujący i obserwuje się zmniejszenie krzepliwości krwi.
Bez niezbędnej terapii objawy nasilają się w miarę odurzenia organizmu dziecka i wzrasta ryzyko dla prawidłowego rozwoju, a nawet życia człowieka. 20-30% dzieci w przypadku odmowy leczenia lub późnego rozpoznania choroby umiera z powodu sepsy, która rozwija się na skutek aktywności leukocytów. Ci, którzy przeżyją, cierpią na niewydolność nerek i problemy z rozwojem psychomotorycznym.
Pod wieloma względami stopień manifestacji patologii zależy od przyczyny dziedzicznej. Galaktozemia objawia się niedoborem GALT. Tak więc objaw Duharta na początku może objawiać się jedynie przedłużającą się żółtaczką (do 2 miesięcy po urodzeniu), nieco później zaćma i zaburzenia czynności wątroby. Jeśli jednak aktywność enzymu GALT wynosi co najmniej 50%, objawy kliniczne mogą nie pojawić się.
Brak enzymu GALA nie jest aż tak wyraźny. Głównym objawem choroby jest postępująca zaćma, a z czasem w moczu mogą pojawiać się także monosacharydy. Jednocześnie utrata masy ciała dziecka jest niewielka, a zdolności psychomotoryczne rozwijają się w normalnym zakresie.
Niedobór GALE początkowo występuje niezwykle rzadko, jednak objawy nadal dzielą się na dwie postacie: łagodną, gdy brak enzymu wpływa na komórki krwi, oraz uogólnioną, gdy niedobór wpływa na wszystkie tkanki organizmu. W pierwszym przypadku jedynie analiza galaktozemii pomoże wykryć chorobę we wczesnych stadiach, dopiero z czasem można zauważyć dysfunkcję zdolności psychomotorycznych i rozwoju mowy. Druga postać w swoich objawach przypomina niedobór GALT, jednak w tym przypadku występuje również powiększenie śledziony. Nawet podczas leczenia ciężkiej postaci choroby GALE, z biegiem czasu u dziecka występuje opóźniony rozwój psychomotoryczny, głuchota i problemy ze wzrokiem.
Rozpoznanie galaktozemii
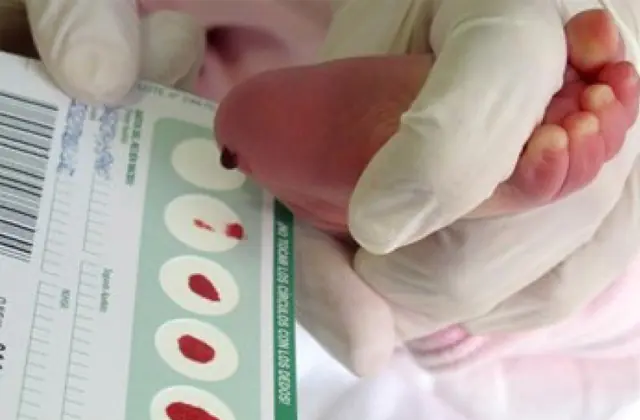
Wprowadzone w wielu krajach badania przesiewowe noworodków pozwalają wykryć we wczesnym stadium nawet 50 chorób wrodzonych, w tym niedostateczną produkcję enzymów biorących udział w metabolizmie. Jeżeli z jakiegoś powodu nie przeprowadzono badań przesiewowych, chorobę rozpoznaje się na podstawie zbiorczych cech charakterystycznych.
Zatem w przypadku podejrzenia galaktozemii zalecenia lekarskie są formułowane dopiero po szczegółowej diagnozie. Mały pacjent musi przejść:
- badanie ogólne - czy występują oznaki żółtaczki;
- ocenia się objawy ogólne - ilość i jakość wypróżnień, obecność lub brak hipotoniczności mięśni, problemy z umiejętnościami psychomotorycznymi;
- badania laboratoryjne - badanie krwi na biochemię, ogólne badanie krwi, badanie moczu na obecność cukru i białka;
- dodatkowe badanie stanu wątroby i nerek.
Diagnostyka DNA jest obowiązkowa w celu wykrycia mutacji GALT. Jeżeli u dziecka występują objawy choroby, a aktywność enzymów GALT według wyników badań jest prawidłowa, przeprowadza się diagnostykę DNA na obecność enzymu GALE.
Lekarze mogą przepisać szereg dodatkowych badań w celu zróżnicowania diagnozy. Konieczna jest ekskluzywna diagnoza, aby potwierdzić lub obalić obecność cukrzycy i innych patologii związanych z wysokim poziomem cukru, a także chorób powodujących powiększenie wątroby.
Jak leczyć galaktozemię?

Gdy badania przesiewowe noworodków i inne badania potwierdzą obecność galaktozemii, przepisuje się leczenie. Należy zrozumieć, że ponieważ choroba jest spowodowana predyspozycjami genetycznymi, całkowite wyleczenie na tym etapie rozwoju medycyny jest niemożliwe. Terapia ma na celu zmniejszenie objawów klinicznych patologii, minimalizację postępu choroby i rozwoju powikłań.
Główną metodą leczenia jest prawidłowe odżywianie w przypadku galaktozemii. Wszystkie produkty zawierające mleko, nawet chleb, słodycze, wędliny i inne, są całkowicie wyłączone z diety pacjenta. Usuwane są także produkty pochodzenia roślinnego i zwierzęcego zawierające oligosacharydy: rośliny strączkowe, soja, szpinak, czekolada, orzechy, wątroba, mózgi, jaja i pochodne tych produktów. Jedynym sposobem zapobiegania zatruciu organizmu jest rygorystyczna terapia dietetyczna.
W przypadku noworodków specjalistyczne jest również żywienie galaktozemii. Mleko matki jest stopniowo całkowicie eliminowane z diety i zastępowane preparatami bez laktozy oraz preparatami na bazie izolatu białka sojowego. Objętość składników odżywczych w tym przypadku powinna odpowiadać normom dla zdrowego dziecka. Jeśli u dziecka występują objawy alergii na izolat białka sojowego, wybierane są mieszanki na bazie hydrolizatów kazeiny. Pierwsza porcja mieszanki pokarmowej powinna stanowić 1/10 dziennego zapotrzebowania dziecka na składniki odżywcze. W ciągu 5-7 dni mleko matki jest całkowicie eliminowane z diety.
Pierwsze pokarmy uzupełniające dla dzieci z patologią są przepisywane od 4 miesięcy. Dietę uzupełniają soki owocowe (jabłkowy, gruszkowy i inne). Po pół miesiąca wprowadzane są przeciery owocowe. Do 1 roku objętość soku i przecieru spożywanego dziennie wzrasta do 30-50 ml. Pierwszy przecier warzywny w wodzie (z wyjątkiem roślin strączkowych) wprowadza się po 5 miesiącach. Od 5,5 miesiąca dziecku można podawać komercyjne płatki zbożowe bezmleczne (kasza gryczana, ryż, kukurydza). Dziecko może próbować produktów mięsnych od 6 miesiąca życia, preferowane są specjalistyczne konserwy niezawierające mleka ani produktów zawierających mleko.
Wybierając specjalistyczne produkty spożywcze dla niemowląt, należy uważnie czytać etykiety. Za bezpieczne dla dziecka uważa się produkty zawierające nie więcej niż 5 mg galaktozy na 100 g. Jeżeli zawartość galaktozy przekracza 20 mg, podawanie produktu dziecku jest surowo zabronione.
Skuteczność leczenia galaktozemii sprawdzana jest w drodze badania kontrolnego raz na kwartał. Badana jest także skuteczność leczenia objawów towarzyszących patologii.
Ważny! Szczególną uwagę zwraca się na dobór leków dla takich pacjentów. Niektóre leki zawierają w swoim składzie laktozę, inne mogą hamować usuwanie produktów przemiany materii z wątroby, co na ogół pogarsza objawy wady wrodzonej i jest niedopuszczalne w ramach właściwego leczenia.Zapobieganie galaktozemii

Nie opracowano jeszcze specjalistycznych środków zapobiegawczych przeciwko tej chorobie. Jeżeli u rodziców stwierdzono predyspozycję do patologii, zaleca się zaplanowanie poczęcia z dodatkowymi konsultacjami z genetykiem. W czasie ciąży należy także poddać się serii badań (biopsja kosmówki w 10-12 tygodniu i badanie płynu owodniowego w 15-18 tygodniu) w celu wykrycia mutacji w genach GALT i GALE.
Pomimo opracowanego dość skutecznego leczenia objawowego patologii, długoterminowe perspektywy rozwoju dziecka są trudne do przewidzenia. Tym samym wskaźniki rozwoju fizycznego pacjentki będą niższe w porównaniu z rówieśnikami, prawdopodobne jest wystąpienie zaburzeń mowy i koordynacji, zwiększenie łamliwości kości, a u dziewcząt – dysfunkcja jajników.
Od 5. roku życia dziecku można zalecić przyjmowanie witamin i leków zawierających ATP, aby zapobiec postępowi choroby. Od 12. roku życia dziewczętom przepisuje się terapię hormonalną w celu kompensacji dysfunkcji jajników.
Galaktozemia jest dość rzadką chorobą uwarunkowaną genetycznie. Mechanizm rozwoju patologii nie został jeszcze dokładnie zbadany, jednak powaga problemu doprowadziła do opracowania szeregu skutecznych technik diagnostycznych. Badania przesiewowe noworodków, obowiązkowe w wielu krajach, umożliwiają wykrycie choroby na czas i podjęcie odpowiednich działań. I choć leczenie chorób uwarunkowanych genetycznie nie zostało dotychczas w pełni rozwinięte, terapia pomaga pozbyć się ciężkich objawów choroby i jej postępu. Zadaniem rodziców w tym przypadku jest jak najwcześniejsze zdiagnozowanie patologii i przestrzeganie zaleceń lekarza, pomagając w ten sposób dziecku poradzić sobie z wadą wrodzoną.
Co to jest galaktozemia - obejrzyj wideo:
