
Todas as pessoas envelhecem. Segundo os cientistas, se não fossem os efeitos destrutivos do ambiente externo e nossos vícios em prazeres prejudiciais ao corpo, viveríamos até 130, ou até 150 anos. E há 16 anos, em 29 de agosto de 2001, os cientistas chegaram a anunciar que haviam encontrado um gene da longevidade. Então, talvez, num futuro próximo, seremos capazes de viver toda a vida que a natureza nos atribui. Mas, por enquanto, estamos envelhecendo e morrendo, a grande maioria antes dos 80-90 anos. E algumas doenças encurtam várias vezes esse período já não tão longo. E o mais “assassino” entre eles, no sentido literal da palavra, é a progéria. MedAboutMe descobriu como é envelhecer em uma década e meia a duas décadas.
O que é envelhecimento?

O envelhecimento é um processo natural característico de todos os organismos vivos da Terra. Todas as teorias existentes sobre o tema “Por que as pessoas envelhecem?” podem ser divididos em dois grandes grupos. Os proponentes de um deles argumentam que o envelhecimento foi planejado pela natureza para a evolução posterior das espécies e da sociedade. Outros têm certeza de que não há planos globais aqui - simplesmente danos no nível genético e celular se acumulam com o tempo, o que leva ao desgaste do corpo.
De uma forma ou de outra, ao longo da vida de uma pessoa, os resultados de falhas e erros internos, bem como as consequências das influências externas, acumulam-se em suas células e tecidos. Entre os principais fatores do envelhecimento estão os seguintes:
- Exposição a espécies reativas de oxigênio (ROS), que, claro, nosso corpo necessita, mas nem sempre e nem em todos os lugares.
- Mutações no DNA das células somáticas (isto é, células do corpo). O genoma não é uma estrutura congelada no tempo e no espaço. É uma estrutura viva e mutável.
- Acúmulo de proteínas danificadas, que são um subproduto de ERO ou interrupções nos processos metabólicos.
- Encurtamento dos telômeros – as extremidades dos cromossomos. É verdade que recentemente os cientistas começaram a duvidar que o envelhecimento esteja associado aos telômeros, mas até agora essa teoria ainda é popular.
A progéria, que será discutida neste artigo, não é envelhecimento - no sentido em que a ciência a entende quando fala sobre expectativa de vida, desgaste do corpo, etc. uma doença genética associada à produção prejudicada de certas proteínas.
Progéria - doenças de crianças e adultos
O inglês Jonathan Hutchinson, em 1886, descreveu pela primeira vez uma criança de 6 anos na qual observou atrofia da pele. O nome da doença incomum (da palavra grega “progeros” - antiga antes do tempo) foi dado a ela em 1897 pelo Dr. Guilford, que estava estudando e descrevendo as nuances da doença. Em 1904, o Dr. Werner publicou uma descrição da progéria em adultos usando o exemplo de quatro irmãos que sofriam de catarata e esclerodermia.
Acredita-se que Francis Scott Fitzgerald escreveu sua história “O Curioso Caso de Benjamin Button” em 1922 justamente sob a impressão de informações sobre pacientes com progéria. Em 2008, Brad Pitt interpretou o protagonista do livro no filme O Curioso Caso de Benjamin Button.
Existem dois tipos de progéria:
- Uma doença que afeta crianças é a síndrome de Hutchinson-Gilford.
Esta é uma patologia rara. Ocorre em 1 criança em vários milhões. Acredita-se que no mundo hoje não existam mais de cem pessoas que sofrem de progéria infantil. No entanto, os cientistas sugerem que podemos falar de mais 150 casos não diagnosticados.
- Uma doença que afeta adultos é a síndrome de Werner.
Esta também é uma doença rara, mas não tão rara quanto a progéria infantil. Pessoas com síndrome de Werner nascem em 1 caso em 100 mil. No Japão - com um pouco mais de frequência: 1 caso em 20-40 mil recém-nascidos. No total, são conhecidos pouco menos de 1,5 mil desses pacientes no mundo.
Genes e "envelhecimento"

A progéria infantil está apenas indiretamente relacionada ao envelhecimento real. Esta é uma doença do grupo das laminopatias - doenças que se desenvolvem no contexto de um problema com a produção da proteína lamina A. Se não for suficiente, ou se o corpo produzir a lamina A “errada”, então uma de um todo desenvolve-se uma lista de doenças, que inclui a síndrome de Hutchinson-Gilford.
A causa da progéria infantil é uma mutação no gene LMNA, localizado no cromossomo 1. Esse gene codifica o composto prelamina A, do qual se obtém a proteína lamina A, que forma uma placa fina - a lâmina - que cobre a membrana interna do núcleo. É necessário para ancorar todos os tipos de moléculas e estruturas internas do núcleo. Se faltar a lâmina A, a estrutura interna do núcleo da célula não pode ser construída, não consegue manter a estabilidade, o que leva à destruição acelerada das células e de todo o organismo. Além disso, a lâmina A desempenha um papel fundamental na divisão celular. Regula a quebra e restauração do núcleo celular. Não é difícil imaginar o que pode acontecer se essa proteína faltar ou não for o que deveria ser. A mutação do gene LMNA leva à formação da proteína “errada” - a progerina. É isso que provoca o “envelhecimento” acelerado das crianças.
De acordo com os dados mais recentes, a mutação ocorre nos estágios iniciais do desenvolvimento embrionário e quase nunca é transmitida de pai para filho.
Há vários anos, os cientistas descobriram que a progerina também é produzida por células saudáveis, mas em quantidades significativamente menores do que na síndrome de Hutchinson-Gilford. Além disso, descobriu-se que, com a idade, a progerina aumenta nas células normais. E esta é a única coisa que realmente liga a progéria infantil ao processo de envelhecimento.
A progéria adulta é o resultado de outra mutação, no gene WRN. Este gene codifica uma proteína necessária para manter os cromossomos em estado estável e também está envolvido nos processos de divisão celular. Se houver uma mutação no gene WRN, a estrutura dos cromossomos muda constantemente. A frequência de mutações espontâneas aumenta 10 vezes, enquanto a capacidade de divisão das células diminui 3-5 vezes em comparação com células saudáveis. O comprimento dos telômeros também diminui. E esses processos já estão muito próximos do envelhecimento a que nos referimos quando olhamos para os idosos num banco.
Progéria em crianças

Nas crianças, a doença se manifesta pela primeira vez após um ou dois anos. Esses bebês têm baixa estatura inadequada para a idade, cabeça grande com parte facial caracteristicamente reduzida, nariz fino que parece um bico e orelhas salientes. Os sinais característicos incluem micrognatia (maxilar inferior subdesenvolvido) e exoftalmia (olhos esbugalhados). Os progéricos têm uma camada muito fina de tecido adiposo subcutâneo e pele fina e hiperpigmentada - seca rapidamente e fica enrugada, parece translúcida, flácida, com vasos sanguíneos translúcidos. Suas unhas não estão totalmente desenvolvidas ou totalmente ausentes. O cabelo rapidamente fica grisalho e cai, até os cílios e as sobrancelhas ficam esparsos e finos. Tudo isso lhes dá a aparência de velhinhos de aparência característica. Daí o mito sobre as pessoas que envelhecem rapidamente.
Pacientes com progéria geralmente desenvolvem atraso no desenvolvimento sexual e os exames mostram níveis elevados de colesterol e lipoproteínas no sangue. Mas a inteligência dos progéricos não sofre. A esperança média de vida dessas crianças é de 13 anos. Um homem com progéria infantil que viveu 27 anos tornou-se um “fígado longo”.
Algumas pessoas com progéria infantil realizaram mais em suas curtas vidas do que muitos de nós. Leon Botha é um dos progéricos longevos: nos 26 anos que lhe foram atribuídos, conseguiu se tornar um famoso artista, músico e DJ popular. E Sam Burns, dos Estados Unidos, falecido em 2014 aos 17 anos, ficou famoso como palestrante motivacional e ativista na área de divulgação de informações sobre sua doença.
A principal causa de morte em pessoas com síndrome de Hutchinson-Gilford é a aterosclerose, que afeta todo o corpo, e a fibrose miocárdica, a substituição do músculo cardíaco por tecido conjuntivo. Nesse caso, uma substância semelhante à gordura se deposita no cérebro, rins, fígado, ovários (nas mulheres) e testículos (nos homens), e os ossos ficam mais finos.
Progéria em adultos
Em adultos, a síndrome de Werner aparece pela primeira vez entre os 14 e os 18 anos de idade. Eles também experimentam uma diminuição acentuada de peso, o crescimento pára, aparecem cabelos grisalhos e ocorre calvície. Seu corpo está se deteriorando rapidamente: são observados distúrbios no funcionamento dos rins, fígado e glândulas endócrinas, desenvolvimento de osteoporose, aterosclerose, diabetes mellitus, catarata e infarto do miocárdio. Homens e mulheres experimentam mudanças no sistema reprodutivo, o que leva à infertilidade. Cada décima pessoa com progéria adulta é diagnosticada com um tumor maligno aos 30-40 anos de idade - câncer de mama, melanoma, sarcoma, etc. Esses pacientes vivem até cerca de 45-50 anos de idade e morrem de câncer ou doença cardíaca.
Ao contrário da progéria infantil, com a síndrome de Werner a mente do paciente também sofre.
Em busca da cura para o envelhecimento precoce

Ainda não há cura para a progéria. Os médicos estão fazendo o possível para retardar o rápido “envelhecimento” desses pacientes. Crianças com síndrome de Hutchinson-Gilford recebem medicamentos com propriedades antioxidantes, vitamina E, etc. Elas são submetidas à cirurgia de revascularização do miocárdio na tentativa de lidar com complicações no coração e nos vasos sanguíneos.
Um medicamento que pode prolongar a vida de um paciente com progéria infantil em um ano, ou na melhor das hipóteses, em 6 a 7 anos, é o lonafarnib, um medicamento criado para tratar o câncer. Para uma pessoa cuja vida pode terminar entre 13 e 17 anos, este é um período significativo. Em 2012, descobriu-se que tomá-lo leva à melhora da audição, ossos mais fortes, aumento do peso corporal e aumento da flexibilidade vascular. 6 anos não é tanto tempo, mas a criação de muitos medicamentos eficazes começou com conquistas semelhantes.
Resultados encorajadores também foram obtidos com a terapia com os seguintes medicamentos:
rapamicina, um imunossupressor que prolonga a vida dos músculos de crianças com progéria;
pravastatina, que reduz os níveis de colesterol;
zoledronato, que retarda o desenvolvimento de metástases ósseas no câncer.
A prevenção da progéria é necessária? Anteriormente pensava-se que não, uma vez que as pessoas com progéria infantil não viviam o suficiente para poderem dar à luz filhos e os pacientes adultos desenvolviam infertilidade. É verdade que nos últimos anos houve relatos de mulheres com progéria que deram à luz crianças saudáveis. Mas esta doença é tão rara que a probabilidade de propagação de mutações é extremamente baixa.
Nenhuma cura foi encontrada ainda para a progéria em adultos. Nesses pacientes, apenas as doenças graves e numerosas que os atacam são tratadas.
A ciência não fica parada, principalmente no campo da medicina. Nossos ancestrais nem imaginavam que no futuro doenças como a tuberculose (tuberculose) ou a peste se tornariam curáveis. Mas mesmo com esse progresso, existem 15 mil doenças no mundo moderno. E embora uma pessoa possa lidar com muitas doenças virais ou infecciosas, algumas mutações genéticas não podem ser curadas. Apresentamos a você as 10 doenças mais incomuns do mundo.
1. Progéria ou síndrome do envelhecimento prematuro
- Número de pessoas diagnosticadas com a doença:

Criança com doença de progéria
Um ano na vida de uma pessoa assim pode ser considerado 10-15 anos na vida de uma pessoa comum, portanto, em 90% dos casos, as crianças morrem desta doença antes de completar 15 anos de idade. As causas da morte geralmente são ataques cardíacos ou hemorragias múltiplas.
2. Síndrome de Kleine-Levin
- Número de pessoas diagnosticadas com a doença: 20 em todo o mundo
- Esperança média de vida: igual à das pessoas normais
- Razões: desconhecido
- Tratamento: incurável
Na síndrome de Kleine-Levin (também conhecida como “Doença da Bela Adormecida”), os pacientes apresentam periodicamente crises de sono excessivo (de 18 horas por dia), com duração de 2 a 40 dias, que interrompem apenas para satisfazer necessidades biológicas.
É extremamente difícil acordar tal pessoa durante o período de “hibernação” e após acordar ela se torna extremamente agressiva. Ao retornar ao ritmo normal de vida, o paciente experimenta amnésia pelos acontecimentos durante a crise. Em 90% dos casos ocorre em adolescentes e é incurável, embora possa desaparecer espontaneamente com a idade.
3. Fibrodisplasia ou síndrome do homem de pedra
- Número de pessoas diagnosticadas com a doença: 500 em todo o mundo
- Esperança média de vida: 30 anos
- Causa: mutações no nível do gene
- Tratamento: incurável
Até os hematomas são perigosos para as pessoas com fibrodisplasia: no local da lesão (pancada, hematoma ou hematoma), o paciente experimenta a formação de um novo tecido ósseo, um “segundo esqueleto”.
Danos frequentes ao tecido muscular podem levar à imobilização completa de uma pessoa. A medicina moderna não inventou métodos para tratar esta doença, mas os cientistas americanos conduziram uma série de testes bem-sucedidos que dão aos pacientes esperança de um futuro saudável.
4. Porfiria
- Causa: perturbação na síntese de glóbulos vermelhos, doença hereditária
- Tratamento: uso de medicamentos hormonais, hepatoprotetores, transfusão de hemácias.
A porfiria, ou vampirismo, é uma doença genética na qual a pele de uma pessoa, ao entrar em contato com a luz solar, começa a coçar, ficar com cicatrizes, bolhas e estourar.

Mulher com síndrome de vampirismo (porfiria)
Mas a doença não afeta apenas a pele: as unhas, gengivas e orelhas dos pacientes ficam deformadas; Os dentes e as gengivas ficam vermelhos quando expostos ao sol e a córnea dos olhos fica turva. Pessoas com porfiria preferem sair de casa ao entardecer ou à noite, quando não há sol. Talvez por isso surgiram lendas sobre vampiros.
5. Síndrome de Mobius
- Causas: desconhecidas, a doença se baseia na paralisia ou subdesenvolvimento do nervo facial.
- Tratamento: não existem medidas radicais para curar esta síndrome, desde a primeira infância os pacientes são submetidos à correção do estrabismo, da fala, etc. Os defeitos mais pronunciados são eliminados por meio de cirurgia.

Menina com síndrome de Mobius
Esta anomalia congênita é caracterizada pelo desenvolvimento anormal ou paralisia do nervo facial, resultando na ausência total ou parcial de expressões faciais no rosto da pessoa. Pessoas com essa síndrome não conseguem rir e até têm dificuldade para engolir; seu rosto mais parece uma máscara.
6. Epidermólise bolhosa ou doença das borboletas
- Número de pessoas diagnosticadas com a doença: desconhecido
- Esperança média de vida: 30 anos
- Causa: mutação cromossômica
- Tratamento: incurável
Natação, jogos esportivos, abraços - essa é uma pequena lista de coisas inacessíveis ao “gente borboleta”.

Epidermólise bolhosa ou doença das borboletas
A epidermólise bolhosa é caracterizada pela alta sensibilidade da pele, devido à qual em pacientes com o menor dano mecânico (mesmo atrito) na pele, formam-se bolhas e úlceras no local da lesão, causando fortes dores à pessoa.
A doença em si não é fatal, mas 90% das crianças com menos de 3 anos morrem devido a cuidados inadequados e as restantes têm de suportar dores terríveis pelo resto da vida, uma vez que a doença é incurável.
7. Hipertricose ou síndrome do lobo
- Causas: mutações genéticas/disfunção das glândulas endócrinas ou desequilíbrio hormonal
- Tratamento: a forma congênita da doença é incurável; na forma adquirida o tratamento é feito por endocrinologista,

Hipertricose ou síndrome do lobo
Esta doença é típica de crianças em idade precoce e é caracterizada pelo crescimento excessivo de pelos em diferentes partes do corpo que não correspondem à idade/sexo da pessoa. Esse crescimento anormal de pelos faz as pessoas parecerem animais. A hipertricose congênita não faz mal à saúde, mas também não tem tratamento. Os pacientes têm que recorrer constantemente aos serviços de depilação, pois essas violações parecem inestéticas.
8. Elefantíase ou elefantíase
- Causas: parasitas portadores de filariose/desenvolvimento anormal do sistema linfático
- Tratamento: Antibióticos com avermectina.
Com esta doença, devido à estagnação da linfa nos membros e à violação de suas propriedades, um novo tecido conjuntivo é formado no local do edema e, como resultado, o membro aumenta de tamanho várias vezes.
Os membros com elefantíase se assemelham aos elefantinos e as áreas afetadas da pele ficam cobertas de rachaduras, úlceras e verrugas. Essas mudanças impedem que uma pessoa se mova normalmente, pratique atividades vigorosas e até mesmo se lave. A doença é tratável com novos antibióticos descobertos em 2015.
9. Urticária aquagênica ou alergia à água
- Número de pessoas diagnosticadas com a doença: 50 em todo o mundo
- Razões: desconhecido
- Tratamento: incurável; os sintomas são aliviados com anti-histamínicos
Alergia à água ou urticária aquagênica
Ao entrar em contato com a água, a pele do doente sente dor e aparecem erupções cutâneas nas áreas afetadas, acompanhadas de coceira e descamação. Esta doença não é fatal, mas limita muito a vida dos doentes: ocorre uma reação alérgica à chuva, às lágrimas e até ao suor.
10. Síndrome de Cotard ou síndrome do cadáver vivo
- Causas: depressão, transtornos mentais
- Tratamento: uso de antidepressivos e psicotrópicos, observação por psiquiatra
As pessoas que sofrem desta síndrome estão convencidas de que estão mortas e são apenas cadáveres vivos. Eles sentem o cheiro da podridão de seus órgãos internos e de como os vermes os comem por dentro. Os pacientes estão confiantes de que não precisam de comida ou água. Este distúrbio é acompanhado por depressão grave e tendências suicidas.
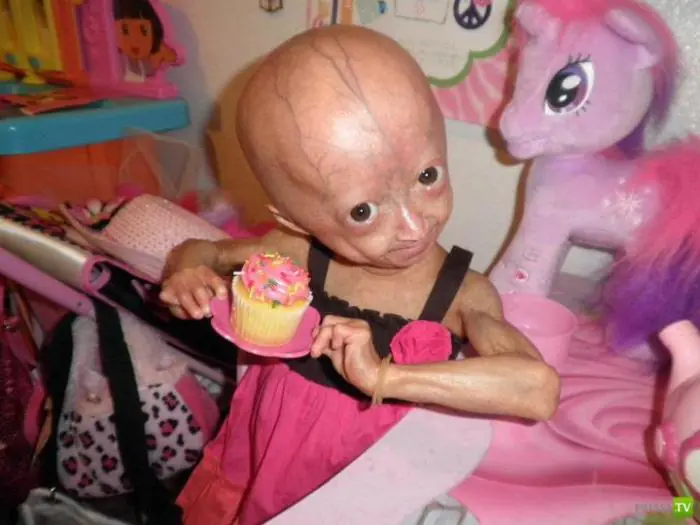
Em outubro de 2005, em uma clínica de Moscou, os médicos realizaram a primeira operação em um paciente que sofria da síndrome do envelhecimento prematuro. A progéria é uma doença muito rara. Os luminares médicos de todo o mundo afirmam que a partir do momento em que esta doença “desperta” no corpo, as pessoas vivem em média apenas 13 anos.
Segundo as estatísticas, aproximadamente 1 em cada 4 milhões de pessoas nascem com esse defeito genético. A progéria é dividida em progéria infantil, chamada síndrome de Hutchinson-Gilford, e progéria adulta, chamada síndrome de Werner. Em ambos os casos, o mecanismo genético entra em colapso e começa um esgotamento não natural de todos os sistemas de suporte à vida. Com a síndrome de Hutchinson-Gilford, o desenvolvimento físico das crianças é atrasado, ao mesmo tempo que aparecem sinais de envelhecimento senil, calvície e rugas nos primeiros meses de vida.
Aos cinco anos, essa criança sofre de todas as doenças da velhice: perda auditiva, artrite, aterosclerose e não chega a viver até os 13 anos. Com a síndrome de Werner, os jovens começam a envelhecer rapidamente aos 16-20 anos e, aos 30-40 anos, esses pacientes morrem com todos os sintomas da velhice extrema.
Não há cura para a progéria - usando todos os avanços científicos, você só pode retardar o processo irreversível.
Juventude roubada
Os casos de envelhecimento repentino são muito prosaicos: uma criança que vive em condições normais surpreende inicialmente as pessoas ao seu redor com seu rápido desenvolvimento. Em tenra idade, ele parece um adulto, e então começa a mostrar todos os sinais de... aproximação da velhice.

Em 1716, na cidade inglesa de Nottingham, morreu o filho de dezoito anos do conde William de Sheffield, que começou a envelhecer aos treze anos. O jovem Sheffield parecia muito mais velho que o pai: cabelos grisalhos, dentes meio perdidos, pele enrugada. O infeliz jovem tinha a aparência de um homem castigado pela vida, sofreu muito com isso e aceitou a morte como libertação do tormento.
Existem casos deste tipo entre representantes de famílias reais. O rei húngaro Ludwig II, aos nove anos, já havia atingido a puberdade e gostava de se divertir com as moças da corte. Aos quatorze anos, adquiriu uma barba espessa e cheia e começou a aparentar ter pelo menos 35 anos. Um ano depois ele se casou e, em seu aniversário de dezesseis anos, sua esposa lhe deu um filho. Mas, aos dezoito anos, Ludwig ficou completamente grisalho e dois anos depois morreu com todos os sinais de decrepitude senil.
É curioso que nem o filho do rei nem os seus descendentes tenham herdado tal doença. Entre os exemplos do século XIX, destaca-se a história de uma simples aldeã, a francesa Louise Ravaillac. Aos oito anos, Louise, já plenamente formada como mulher, engravidou de um pastor local e deu à luz uma criança completamente saudável. Aos 16 anos já tinha três filhos e parecia mais velha que a mãe; aos 25 anos tornou-se uma velha decrépita e, antes de completar 26 anos, morreu de velhice.
Não menos interessante é o destino daqueles que viveram no século XX. Alguns deles tiveram um pouco mais de sorte do que outros. Por exemplo, Michael Sommers, morador da cidade americana de San Bernardino, nascido em 1905, amadureceu e envelheceu cedo, podendo viver até os 31 anos. No início, a entrada super rápida na idade adulta até o agradou. Mas quando, aos dezessete anos, Michael percebeu com horror que havia começado a envelhecer, começou a fazer tentativas desesperadas de interromper esse processo destrutivo.
Mas os médicos apenas encolheram os ombros, incapazes de fazer algo para ajudar. Sommers conseguiu desacelerar um pouco sua decrepitude depois que se mudou definitivamente para a aldeia e começou a passar muito tempo ao ar livre. Mesmo assim, aos 30 anos, ele se tornou um homem velho e, um ano depois, foi liquidado por uma gripe comum. Entre outros fenômenos semelhantes, destaca-se a inglesa Barbara Dahlin, falecida em 1982, aos 26 anos.
Aos 20 anos, casada e tendo dois filhos, Bárbara envelheceu rápida e irreversivelmente. É por isso que seu jovem marido a abandonou, que não queria conviver com a “velha ruína”. Aos 22 anos, devido à deterioração do estado de saúde e aos choques sofridos, a “velha” ficou cega e até à sua morte locomovia-se pelo toque ou acompanhada por um cão-guia, que lhe foi entregue pelas autoridades da sua cidade natal, Birmingham.
Paul Demongeau, da cidade francesa de Marselha, tem 23 anos. Ao mesmo tempo, ele parece ter 60 anos e se sente um velho. No entanto, ele ainda não perdeu a esperança de que um milagre aconteça e que seja encontrado um remédio que irá parar a sua rápida decrepitude. Seu infeliz irmão, um siciliano da cidade de Siracusa, Mario Termini, não tem nem 20 anos, mas parece ter muito mais de 30. Filho de pais ricos, Termini não se nega nada, conhece belezas locais e lidera um estilo de vida desenfreado.
O que nós temos?
Pessoas “precoces” também viveram em nosso país. Mesmo na época de Ivan, o Terrível, o filho dos boiardos Mikhailov, Vasily, morreu aos 19 anos como um velho decrépito. Em 1968, aos 22 anos, Nikolai Shorikov, trabalhador de uma das fábricas, morreu em Sverdlovsk. Ele começou a envelhecer aos dezesseis anos, o que intrigou muito os médicos. Os luminares da medicina apenas deram de ombros: “Isso não pode ser!”
Tendo envelhecido na idade em que tudo estava apenas começando, Nikolai perdeu todo o interesse pela vida e cometeu suicídio engolindo comprimidos... E treze anos depois, o “velho” Sergei Efimov, de 28 anos, morreu em Leningrado. Seu período de juventude terminou aos onze anos, e ele começou a envelhecer visivelmente depois dos vinte e morreu velho decrépito, tendo perdido quase completamente a capacidade de pensar com sensatez um ano antes de sua morte.
Os genes são os culpados por tudo
Muitos cientistas acreditam que a principal causa desta doença é uma mutação genética que leva ao acúmulo de grandes quantidades de proteínas nas células. Videntes e mágicos afirmam que existem técnicas especiais para enviar “danos” para fazer uma pessoa envelhecer.
Aliás, esta doença ocorre não só em humanos, mas também em animais. Eles também têm ciclos de vida e períodos que às vezes seguem o roteiro de um ano em três ou até dez anos. Talvez uma solução para o problema seja encontrada após muitos anos de experiências com nossos irmãos menores.
Como estabeleceram pesquisadores da Universidade da Califórnia, um medicamento chamado inibidor da farnesiltransferase reduz significativamente a taxa de sintomas de envelhecimento prematuro em ratos de laboratório. Talvez este medicamento seja adequado para o tratamento de pessoas.
Veja como o Candidato em Ciências Biológicas Igor Bykov caracteriza os sintomas da doença em crianças: “A progéria ocorre repentinamente com o aparecimento de grandes manchas pigmentares no corpo. Então as pessoas começam a sofrer de verdadeiras doenças senis. Eles desenvolvem doenças cardíacas, doenças vasculares, diabetes, cabelos e dentes caem e a gordura subcutânea desaparece. Os ossos ficam quebradiços, a pele fica enrugada e os corpos ficam curvados. O processo de envelhecimento nesses pacientes ocorre aproximadamente dez vezes mais rápido do que em uma pessoa saudável. O mal provavelmente está enraizado nos genes. Existe a hipótese de que de repente eles parem de dar o comando para as células se dividirem. E eles rapidamente se tornam inutilizáveis.”
Os genes param de dar o comando às células para se dividirem, aparentemente devido ao fato de que as extremidades do DNA nos cromossomos são encurtadas - os chamados telômeros, cujo comprimento presumivelmente mede a duração de uma vida humana. Processos semelhantes ocorrem em pessoas normais, mas muito mais lentamente. Mas não está completamente claro como resultado de que tipo de distúrbio os telômeros são encurtados e o envelhecimento começa a acelerar pelo menos 10 vezes. Agora os cientistas estão usando enzimas para alongar os telômeros. Houve até relatos de que geneticistas americanos conseguiram prolongar a vida das moscas dessa forma. Mas ainda estamos longe de resultados que possam ser aplicados na prática. As pessoas não podem ser ajudadas nem mesmo no nível dos experimentos. Felizmente, a doença não é hereditária.
Supõe-se que ocorra um mau funcionamento no genoma durante o período de desenvolvimento intrauterino. Até agora, a ciência não consegue monitorizar e gerir esta falha: só pode afirmar um facto, mas talvez num futuro próximo a gerontologia responda a esta questão ao mundo.
