
Wszyscy ludzie się starzeją. Zdaniem naukowców, gdyby nie destrukcyjny wpływ środowiska zewnętrznego i nasze uzależnienie od szkodliwych dla organizmu przyjemności, żylibyśmy nawet 130, a nawet 150 lat. A 16 lat temu, 29 sierpnia 2001 roku, naukowcy ogłosili nawet, że odkryli gen długowieczności. Być może w najbliższej przyszłości będziemy mogli przeżyć cały okres życia przydzielony nam przez naturę. Ale na razie starzejemy się i umieramy, zdecydowana większość przed 80-90 rokiem życia. A niektóre choroby skracają ten i tak już nie tak długi okres kilkukrotnie. A najbardziej „zabójcą” wśród nich, w dosłownym tego słowa znaczeniu, jest progeria. MedAboutMe dowiedziało się, jak to jest zestarzeć się w ciągu półtora do dwóch dekad.
Co to jest starzenie się?

Starzenie się jest naturalnym procesem charakterystycznym dla każdego żywego organizmu na Ziemi. Wszystkie istniejące teorie na temat „Dlaczego ludzie się starzeją?” można podzielić na dwie duże grupy. Zwolennicy jednego z nich argumentują, że starzenie się było zamierzone przez naturę w celu dalszej ewolucji gatunków i społeczeństwa. Inni są pewni, że nie ma tu żadnych globalnych planów – po prostu uszkodzenia na poziomie genowym i komórkowym kumulują się z czasem, co prowadzi do zużycia organizmu.
Tak czy inaczej, w ciągu życia człowieka w jego komórkach i tkankach gromadzą się skutki wewnętrznych niepowodzeń i błędów, a także konsekwencje wpływów zewnętrznych. Do kluczowych czynników starzenia zalicza się:
- Ekspozycja na reaktywne formy tlenu (ROS), których oczywiście nasz organizm potrzebuje, jednak nie zawsze i nie wszędzie.
- Mutacje w DNA komórek somatycznych (czyli komórek ciała). Genom nie jest strukturą zamrożoną w czasie i przestrzeni. Jest to struktura żywa i zmienna.
- Akumulacja uszkodzonych białek, będących produktem ubocznym ROS lub zakłóceń w procesach metabolicznych.
- Skrócenie telomerów – końców chromosomów. To prawda, że ostatnio naukowcy zaczęli wątpić w związek starzenia się z telomerami, ale jak dotąd teoria ta jest nadal popularna.
Progeria, o której będzie mowa w tym artykule, to nie starzenie się – w takim sensie, w jakim rozumie ją nauka, gdy mówi o średniej długości życia, zużyciu organizmu itp. Choroba ta wygląda jak starzenie się, chociaż w rzeczywistości jest ciężka choroba genetyczna związana z upośledzoną produkcją niektórych białek.
Progeria – choroby dzieci i dorosłych
Anglik Jonathan Hutchinson w 1886 roku po raz pierwszy opisał 6-letnie dziecko, u którego zaobserwował zanik skóry. Nazwę niezwykłej choroby (od greckiego słowa „progeros” – stare przed swoimi czasami) nadał jej w 1897 roku dr Guilford, który badał i opisywał niuanse choroby. W 1904 roku dr Werner opublikował opis progerii u dorosłych na przykładzie czwórki rodzeństwa cierpiącego na zaćmę i twardzinę.
Uważa się, że Francis Scott Fitzgerald napisał swoje opowiadanie „Ciekawy przypadek Benjamina Buttona” w 1922 roku właśnie pod wrażeniem informacji o chorych na progerię. W 2008 roku Brad Pitt zagrał głównego bohatera książki w filmie Ciekawy przypadek Benjamina Buttona.
Istnieją dwa rodzaje progerii:
- Chorobą dotykającą dzieci jest zespół Hutchinsona-Gilforda.
To rzadka patologia. Występuje u 1 dziecka na kilka milionów. Uważa się, że w dzisiejszym świecie na progerię dziecięcą cierpi nie więcej niż sto osób. Naukowcy sugerują jednak, że możemy mówić o około 150 niezdiagnozowanych przypadkach więcej.
- Chorobą dotykającą dorosłych jest zespół Wernera.
Jest to również choroba rzadka, ale nie tak rzadka jak progeria dziecięca. Osoby z zespołem Wernera rodzą się w 1 przypadku na 100 tys. W Japonii - nieco częściej: 1 przypadek na 20-40 tysięcy noworodków. W sumie na świecie znanych jest nieco niecałe 1,5 tys. takich pacjentów.
Geny i „starzenie się”

Progeria u dzieci jest jedynie pośrednio powiązana z faktycznym starzeniem się. Jest to choroba z grupy laminopatii – chorób, które rozwijają się na tle problemu z produkcją laminatu A. Jeśli to nie wystarczy lub organizm wytwarza „niewłaściwy” laminat A, to jedna z całości rozwija się lista chorób, do których zalicza się zespół Hutchinsona-Gilforda.
Przyczyną progerii dziecięcej jest mutacja w genie LMNA, który znajduje się na chromosomie 1. Gen ten koduje złożoną prelaminę A, z której otrzymuje się laminę białkową A, która tworzy cienką płytkę – blaszkę – pokrywającą wewnętrzną błonę jądra. Jest niezbędny do zakotwiczenia wszelkiego rodzaju cząsteczek i struktur wewnętrznych jądra. W przypadku braku laminatu A nie można zbudować wewnętrznego szkieletu jądra komórkowego, nie może ono utrzymać stabilności, co prowadzi do przyspieszonego zniszczenia komórek i całego organizmu. Ponadto laminat A odgrywa kluczową rolę w podziale komórek. Reguluje rozkład i odbudowę jądra komórkowego. Nietrudno sobie wyobrazić, co może się stać, jeśli tego białka zabraknie lub nie będzie takie, jakie powinno być. Mutacja genu LMNA prowadzi do powstania „niewłaściwego” białka – progeryny. To właśnie powoduje przyspieszone „starzenie się” dzieci.
Według najnowszych danych mutacja występuje we wczesnych stadiach rozwoju zarodka i prawie nigdy nie jest przenoszona z rodzica na dziecko.
Kilka lat temu naukowcy odkryli, że progeryna jest również wytwarzana przez zdrowe komórki, ale w znacznie mniejszych ilościach niż w przypadku zespołu Hutchinsona-Gilforda. Co więcej, okazało się, że wraz z wiekiem w prawidłowych komórkach wzrasta poziom progeryny. I tylko to tak naprawdę łączy progerię dziecięcą z procesem starzenia.
Dorosła progeria jest wynikiem innej mutacji w genie WRN. Gen ten koduje białko niezbędne do utrzymania chromosomów w stabilnym stanie, a także bierze udział w procesach podziału komórek. Jeśli w genie WRN występuje mutacja, struktura chromosomów stale się zmienia. Częstotliwość spontanicznych mutacji wzrasta 10-krotnie, natomiast zdolność komórek do podziałów zmniejsza się 3-5-krotnie w porównaniu do komórek zdrowych. Zmniejsza się także długość telomerów. A procesy te są już bardzo bliskie starzeniu się, które mamy na myśli, patrząc na starsze osoby na ławce.
Progeria u dzieci

U dzieci choroba objawia się po roku lub dwóch. Dzieci te charakteryzują się nieodpowiednim dla ich wieku niskim wzrostem, dużą głową z charakterystycznie obniżoną częścią twarzową, cienkim nosem przypominającym dziób i odstającymi uszami. Charakterystyczne objawy to mikrognacja (słabo rozwinięta żuchwa) i wytrzeszcz (wyłupiaste oczy). Progeryki mają bardzo cienką warstwę podskórnej tkanki tłuszczowej i cienką przebarwioną skórę - szybko wysycha i staje się pomarszczona, wygląda na półprzezroczystą, zwiotczałą, z przezroczystymi naczyniami krwionośnymi. Ich paznokcie albo nie są w pełni rozwinięte, albo są całkowicie nieobecne. Włosy szybko siwieją i wypadają, nawet rzęsy i brwi są rzadkie i cienkie. Wszystko to nadaje im wygląd małych staruszków o charakterystycznym wyglądzie. Stąd mit o szybko starzejącym się człowieku.
U pacjentów chorych na progerię zwykle rozwija się opóźniony rozwój seksualny, a badania wykazują podwyższony poziom cholesterolu i lipoprotein we krwi. Ale inteligencja progerów nie cierpi. Średnia długość życia takich dzieci wynosi 13 lat. Mężczyzna cierpiący na progerię w dzieciństwie, który żył 27 lat, stał się „długą wątrobą”.
Niektóre osoby cierpiące na progerię dziecięcą osiągnęły w swoim krótkim życiu więcej niż wielu z nas. Leon Botha to jeden z długowiecznych progeryków, w ciągu 26 lat, jakie mu przydzielono, udało mu się zostać sławnym artystą, muzykiem i popularnym DJ-em. Natomiast Sam Burns ze Stanów Zjednoczonych, zmarły w 2014 roku w wieku 17 lat, zasłynął jako mówca motywacyjny i działacz na rzecz rozpowszechniania informacji o swojej chorobie.
Główną przyczyną zgonów osób z zespołem Hutchinsona-Gilforda jest miażdżyca, która atakuje cały organizm, oraz zwłóknienie mięśnia sercowego, czyli zastąpienie mięśnia sercowego tkanką łączną. W tym przypadku substancja tłuszczopodobna odkłada się w mózgu, nerkach, wątrobie, jajnikach (u kobiet) i jądrach (u mężczyzn), a kości stają się cieńsze.
Progeria u dorosłych
U dorosłych zespół Wernera pojawia się po raz pierwszy między 14 a 18 rokiem życia. Doświadczają także gwałtownego spadku masy ciała, zatrzymania wzrostu, pojawienia się siwych włosów i rozwoju łysienia. Ich organizm szybko się pogarsza: obserwuje się zaburzenia w funkcjonowaniu nerek, wątroby i gruczołów dokrewnych, rozwija się osteoporoza, miażdżyca, cukrzyca, zaćma i zawał mięśnia sercowego. Mężczyźni i kobiety doświadczają zmian w układzie rozrodczym, co prowadzi do niepłodności. Co dziesiąta osoba chora na progerię u dorosłych w wieku 30-40 lat diagnozuje nowotwór złośliwy - rak piersi, czerniak, mięsak itp. Pacjenci tacy dożywają około 45-50 lat i umierają na raka lub choroby serca.
W przeciwieństwie do progerii dziecięcej, w przypadku zespołu Wernera cierpi również umysł pacjenta.
W poszukiwaniu leku na przedwczesne starzenie się

Na progerię nie ma jeszcze leku. Lekarze robią wszystko, co w ich mocy, aby spowolnić szybkie „starzenie się” takich pacjentów. Dzieciom z zespołem Hutchinsona-Gilforda przepisuje się leki o właściwościach przeciwutleniających, witaminę E itp. Poddaje się je operacji bajpasów wieńcowych, próbując uporać się z powikłaniami w sercu i naczyniach krwionośnych.
Lekiem, który może przedłużyć życie pacjenta z progerią dziecięcą o rok, a w najlepszym przypadku o 6-7 lat, jest lonafarnib, lek stworzony do leczenia raka. Dla osoby, której życie może zakończyć się w wieku 13-17 lat, jest to znaczący okres. W 2012 roku odkryto, że jego przyjmowanie prowadzi do poprawy słuchu, mocniejszych kości, zwiększenia masy ciała i zwiększenia elastyczności naczyń. 6 lat to niewiele, ale tworzenie wielu skutecznych leków rozpoczęło się od podobnych osiągnięć.
Zachęcające rezultaty uzyskano także w trakcie terapii następującymi lekami:
rapamycyna, lek immunosupresyjny przedłużający życie mięśni dzieci chorych na progerię;
prawastatyna, która obniża poziom cholesterolu;
zoledronian, który spowalnia rozwój przerzutów do kości w przypadku raka.
Czy profilaktyka progerii jest konieczna? Wcześniej sądzono, że nie, gdyż osoby z progerią dziecięcą nie żyły wystarczająco długo, aby móc urodzić dzieci, a u dorosłych pacjentów występowała niepłodność. To prawda, że w ostatnich latach pojawiły się doniesienia o kobietach chorych na progerię rodzących zdrowe dzieci. Choroba ta jest jednak tak rzadka, że prawdopodobieństwo rozprzestrzenienia się mutacji jest niezwykle niskie.
Na progerię u dorosłych nie wynaleziono jeszcze leku. U takich pacjentów leczy się tylko te ciężkie i liczne choroby, które ich atakują.
Nauka nie stoi w miejscu, zwłaszcza w dziedzinie medycyny. Nasi przodkowie nie mogli sobie nawet wyobrazić, że w przyszłości choroby takie jak gruźlica czy dżuma staną się uleczalne. Ale nawet przy takim postępie we współczesnym świecie istnieje 15 000 chorób. I chociaż dana osoba może poradzić sobie z wieloma chorobami wirusowymi lub zakaźnymi, niektórych mutacji genetycznych nie można wyleczyć. Przedstawiamy Państwu 10 najbardziej niezwykłych chorób świata.
1. Progeria, czyli zespół przedwczesnego starzenia się
- Liczba osób, u których zdiagnozowano tę chorobę:

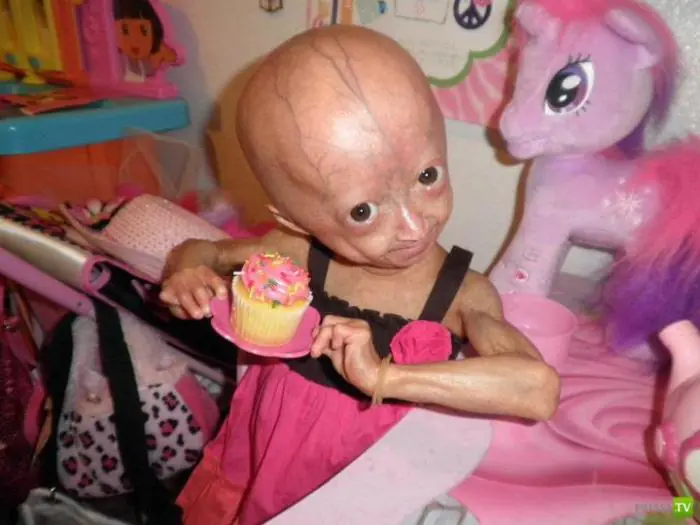
Dziecko chore na progerię
Rok życia takiej osoby można uznać za 10-15 lat życia zwykłego człowieka, dlatego w 90% przypadków dzieci umierają z powodu tej choroby przed ukończeniem 15 roku życia. Przyczyną śmierci są zwykle zawały serca lub liczne krwotoki.
2. Zespół Kleinego-Levina
- Liczba osób, u których zdiagnozowano tę chorobę: 20 na całym świecie
- Średnia długość życia: taka sama jak normalnych ludzi
- Powody: nieznane
- Leczenie: nieuleczalne
W przypadku zespołu Kleinego-Levina (zwanego także „chorobą Śpiącej Królewny”) u pacjentów okresowo występują napady nadmiernego snu (od 18 godzin na dobę), trwające od 2 do 40 dni, które przerywają jedynie w celu zaspokojenia potrzeb biologicznych.
Niezwykle trudno jest obudzić taką osobę w okresie „hibernacji”, a po przebudzeniu staje się ona niezwykle agresywna. Po powrocie do normalnego rytmu życia pacjent doświadcza amnezji związanej z wydarzeniami, które miały miejsce podczas ataku. W 90% przypadków występuje u nastolatków i jest nieuleczalna, choć z wiekiem może samoistnie ustąpić.
3. Fibrodysplazja, czyli zespół kamiennego człowieka
- Liczba osób, u których zdiagnozowano tę chorobę: 500 na całym świecie
- Średnia długość życia: 30 lat
- Przyczyna: mutacje na poziomie genu
- Leczenie: nieuleczalne
Nawet siniaki są niebezpieczne dla osób z fibrodysplazją: w miejscu urazu (uderzenie, krwiak lub siniak) pacjent doświadcza tworzenia się nowej tkanki kostnej, „drugiego szkieletu”.
Częste uszkodzenie tkanki mięśniowej może prowadzić do całkowitego unieruchomienia osoby. Współczesna medycyna nie wynalazła metod leczenia tej choroby, jednak amerykańscy naukowcy przeprowadzili szereg udanych badań, które dają pacjentom nadzieję na zdrową przyszłość.
4. Porfiria
- Przyczyna: zaburzenia w syntezie czerwonych krwinek, choroba dziedziczna
- Leczenie: przyjmowanie leków hormonalnych, hepatoprotektorów, przetaczanie krwinek czerwonych.
Porfiria, czyli wampiryzm, to choroba genetyczna, w której skóra człowieka pod wpływem światła słonecznego zaczyna swędzić, pojawiać się blizn, pojawiać się pęcherze i pękać.

Kobieta z zespołem wampiryzmu (porfiria)
Ale choroba dotyka nie tylko skóry: paznokcie, dziąsła i uszy pacjentów są zdeformowane; Zęby i dziąsła stają się czerwone pod wpływem słońca, a rogówka oczu staje się mętna. Osoby chore na porfirię wolą wychodzić z domu wieczorem lub w nocy, gdy nie ma słońca. Być może z tego powodu pojawiły się legendy o wampirach.
5. Zespół Mobiusa
- Przyczyny: nieznana, choroba polega na porażeniu lub niedorozwoju nerwu twarzowego.
- Leczenie: nie ma radykalnych środków wyleczenia tego zespołu, od wczesnego dzieciństwa pacjenci poddawani są korekcji zeza, mowy itp. Najbardziej widoczne wady są eliminowane operacyjnie.

Dziewczyna z zespołem Mobiusa
Ta wrodzona anomalia charakteryzuje się nieprawidłowym rozwojem lub porażeniem nerwu twarzowego, w wyniku czego mimika jest całkowicie lub częściowo nieobecna na twarzy osoby. Osoby z tym zespołem nie mogą się śmiać, a nawet mają trudności z połykaniem, a ich twarz przypomina bardziej maskę.
6. Pęcherzowe oddzielanie się naskórka, czyli choroba motyli
- Liczba osób, u których zdiagnozowano tę chorobę: nieznana
- Średnia długość życia: 30 lat
- Przyczyna: mutacja chromosomowa
- Leczenie: nieuleczalne
Pływanie, zabawy sportowe, uściski – to krótka lista rzeczy niedostępnych dla „ludzi-motylów”.

Pęcherzowe oddzielanie się naskórka, czyli choroba motyli
Pęcherzowe oddzielanie się naskórka charakteryzuje się dużą wrażliwością skóry, przez co u pacjentów z najmniejszym uszkodzeniem mechanicznym (nawet tarciem) skóry w miejscu urazu tworzą się pęcherze i owrzodzenia, powodując silny ból osoby.
Sama choroba nie jest śmiertelna, ale 90% dzieci poniżej 3 roku życia umiera z powodu niewłaściwej opieki, a reszta musi znosić straszny ból do końca życia, ponieważ choroba jest nieuleczalna.
7. Hipertrychoza, czyli zespół wilka
- Przyczyny: mutacje genów/dysfunkcja gruczołów dokrewnych lub brak równowagi hormonalnej
- Leczenie: wrodzona postać choroby jest nieuleczalna, w postaci nabytej leczenie prowadzi endokrynolog,

Hipertrychoza lub zespół wilka
Choroba ta jest typowa dla dzieci już w młodym wieku i charakteryzuje się nadmiernym owłosieniem na różnych częściach ciała, nie odpowiadającym wiekowi/płci danej osoby. Ten nieprawidłowy wzrost włosów sprawia, że ludzie wyglądają jak zwierzęta. Wrodzone nadmierne owłosienie nie jest groźne dla zdrowia, ale też nie można go leczyć. Pacjenci muszą stale uciekać się do usług usuwania włosów, ponieważ te naruszenia wyglądają nieestetycznie.
8. Słoniowata lub słoniowata
- Przyczyny: pasożyty przenoszące filariozę/nieprawidłowy rozwój układu limfatycznego
- Leczenie: Antybiotyki z awermektyną.
W przypadku tej choroby, z powodu zastoju limfy w kończynach i zakłócenia jej właściwości, w miejscu obrzęku powstaje nowa tkanka łączna, w wyniku czego kończyna kilkakrotnie powiększa się.
Kończyny u słoniowatych przypominają słoniowate, a dotknięte obszary skóry pokryte są pęknięciami, wrzodami i brodawkami. Takie zmiany uniemożliwiają normalne poruszanie się, wykonywanie energicznych czynności, a nawet mycie się. Chorobę można leczyć nowymi antybiotykami odkrytymi w 2015 roku.
9. Pokrzywka wodna, czyli alergia na wodę
- Liczba osób, u których zdiagnozowano tę chorobę: 50 na całym świecie
- Powody: nieznane
- Leczenie: nieuleczalne; objawy ustępują po zastosowaniu leków przeciwhistaminowych
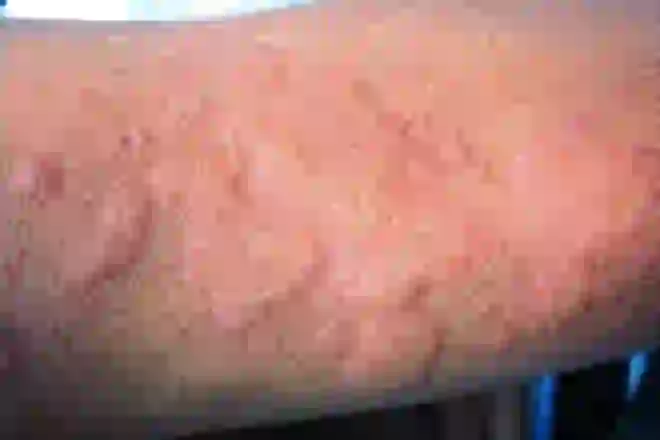
Alergia na wodę lub pokrzywka wodna
W kontakcie z wodą skóra chorego odczuwa ból, a na dotkniętych obszarach pojawiają się wysypki, którym towarzyszy swędzenie i łuszczenie się. Choroba ta nie jest śmiertelna, ale znacznie ogranicza życie chorych: pojawia się reakcja alergiczna na deszcz, łzy, a nawet pot.
10. Zespół Cotarda, czyli syndrom żywego trupa
- Przyczyny: depresja, zaburzenia psychiczne
- Leczenie: przyjmowanie leków przeciwdepresyjnych i psychotropowych, obserwacja psychiatry
Osoby cierpiące na ten syndrom są przekonane, że nie żyją i są tylko żywymi trupami. Czują gnicie ich narządów wewnętrznych i robaki zjadające je od środka. Pacjenci mają pewność, że nie potrzebują jedzenia ani wody. Zaburzeniu temu towarzyszy ciężka depresja i tendencje samobójcze.
W październiku 2005 roku w moskiewskiej klinice lekarze przeprowadzili pierwszą operację u pacjenta cierpiącego na zespół przedwczesnego starzenia się. Progeria to bardzo rzadka choroba. Światowi luminarze medycyny twierdzą, że od momentu „przebudzenia się” tej choroby w organizmie człowiek żyje średnio zaledwie 13 lat.
Według statystyk około 1 na 4 miliony ludzi rodzi się z taką wadą genetyczną. Progerię dzieli się na progerię dziecięcą, zwaną zespołem Hutchinsona-Gilforda, oraz progerię dorosłych, zwaną zespołem Wernera. W obu przypadkach mechanizm genetyczny załamuje się i rozpoczyna się nienaturalne wyczerpywanie się wszystkich systemów podtrzymywania życia. W przypadku zespołu Hutchinsona-Gilforda rozwój fizyczny dzieci jest opóźniony, a jednocześnie w pierwszych miesiącach życia pojawiają się u nich oznaki starczego siwienia, łysienia i zmarszczek.
W wieku pięciu lat takie dziecko cierpi na wszystkie dolegliwości starości: utratę słuchu, zapalenie stawów, miażdżycę i nie dożywa nawet 13 lat. W przypadku zespołu Wernera młodzi ludzie zaczynają szybko się starzeć w wieku 16–20 lat, a w wieku 30–40 lat tacy pacjenci umierają ze wszystkimi objawami skrajnej starości.
Na progerię nie ma lekarstwa – korzystając ze wszelkich zdobyczy nauki, można jedynie spowolnić nieodwracalny proces.
Skradziona młodość
Przypadki nagłego starzenia się są bardzo prozaiczne: dziecko żyjące w normalnych warunkach początkowo zaskakuje otaczających go ludzi swoim szybkim rozwojem. W młodym wieku wygląda jak dorosły, a potem zaczynają wykazywać wszelkie oznaki... zbliżającej się starości.

W 1716 roku w angielskim mieście Nottingham zmarł osiemnastoletni syn hrabiego Williama z Sheffield, który zaczął się starzeć w wieku trzynastu lat. Młody Sheffield wyglądał na znacznie starszego od ojca: siwe włosy, na wpół utracone zęby, pomarszczona skóra. Nieszczęsny młodzieniec wyglądał jak człowiek umęczony życiem, bardzo to wycierpiał i przyjął śmierć jako wybawienie od męk.
Zdarzają się tego typu przypadki wśród przedstawicieli rodzin królewskich. Węgierski król Ludwik II w wieku dziewięciu lat osiągnął już wiek dojrzewania i lubił bawić się z dziewczętami dworskimi. W wieku czternastu lat zapuścił gęstą, pełną brodę i zaczął wyglądać na co najmniej 35 lat. Rok później ożenił się, a w dniu jego szesnastych urodzin żona urodziła mu syna. Ale w wieku osiemnastu lat Ludwig całkowicie posiwiał, a dwa lata później zmarł ze wszystkimi oznakami starczej niedołężności.
Ciekawe, że ani syn króla, ani jego dalsi potomkowie nie odziedziczyli takiej choroby. Wśród przykładów XIX wieku można wyróżnić historię prostej wiejskiej dziewczyny, Francuzki Louise Ravaillac. W wieku ośmiu lat Louise, w pełni uformowana jako kobieta, zaszła w ciążę z miejscowym pasterzem i urodziła całkowicie zdrowe dziecko. Do szesnastych urodzin miała już troje dzieci i wyglądała na starszą od matki; w wieku 25 lat zamieniła się w zniedołężniałą staruszkę i przed ukończeniem 26 lat zmarła ze starości.
Nie mniej interesujące są losy tych, którzy żyli w XX wieku. Niektórzy z nich mieli trochę więcej szczęścia niż inni. Na przykład Michael Sommers, mieszkaniec amerykańskiego miasta San Bernardino, urodzony w 1905 roku, wcześnie dojrzał i postarzał się, dzięki czemu mógł dożyć 31 lat. Początkowo superszybkie wejście w dorosłość nawet go cieszyło. Kiedy jednak w wieku siedemnastu lat Michael z przerażeniem zdał sobie sprawę, że zaczął się starzeć, zaczął desperacko próbować zatrzymać ten destrukcyjny proces.
Ale lekarze tylko wzruszyli ramionami, nie mogąc nic zrobić, aby pomóc. Sommersowi udało się nieco spowolnić swoje zniedołężnienie, gdy przeprowadził się na stałe do wioski i zaczął spędzać dużo czasu na świeżym powietrzu. Ale mimo to w wieku 30 lat stał się starym człowiekiem, a rok później wykończyła go zwykła grypa. Wśród innych podobnych zjawisk można wymienić Angielkę Barbarę Dahlin, która zmarła w 1982 roku w wieku 26 lat.
W wieku 20 lat, po ślubie i urodzeniu dwójki dzieci, Barbara szybko i nieodwracalnie się postarzała. Dlatego opuścił ją młody mąż, który nie chciał żyć ze „starym wrakiem”. W wieku 22 lat na skutek pogarszającego się stanu zdrowia i wstrząsów, jakie przeżyła, „staruszka” straciła wzrok i aż do śmierci poruszała się za pomocą dotyku lub w towarzystwie psa przewodnika, podarowanego jej przez władze rodzinnego Birmingham.
Paul Demongeau z francuskiej Marsylii ma dwadzieścia trzy lata. Jednocześnie wygląda na 60 lat i czuje się jak starzec. Jednak nie stracił jeszcze nadziei, że stanie się cud i zostanie znalezione lekarstwo, które powstrzyma jego szybkie zniedołężnienie. Jego brat w nieszczęściu, Sycylijczyk z miasta Syrakuzy, Mario Termini, nie ma nawet 20 lat, ale wygląda na znacznie więcej niż 30. Syn zamożnych rodziców, Termini niczego sobie nie odmawia, spotyka się z lokalnymi pięknościami i prowadzi burzliwy tryb życia.
Co my mamy?
W naszym kraju mieszkali także ludzie „przedwcześnie rozwinięci”. Już za czasów Iwana Groźnego syn bojarów Michajłowa, Wasilij, zmarł w wieku 19 lat jako zgrzybiały starzec. W 1968 roku w Swierdłowsku zmarł w wieku 22 lat Nikołaj Szorikow, pracownik jednej z fabryk. Zaczął się starzeć w wieku szesnastu lat, co bardzo zaskoczyło lekarzy. Luminanci medycyny tylko wzruszyli ramionami: „To nie może być!”
Stając się starym człowiekiem w wieku, w którym wszystko dopiero się zaczyna, Mikołaj stracił całe zainteresowanie życiem i popełnił samobójstwo, połykając pigułki... A trzynaście lat później 28-letni „starzec” Siergiej Efimow zmarł w Leningradzie. Jego młodzieńczy okres zakończył się w wieku jedenastu lat, a po dwudziestce zaczął się zauważalnie starzeć i zmarł jako zgrzybiały starzec, prawie całkowicie tracąc zdolność rozsądnego myślenia na rok przed śmiercią.
Za wszystko winne są geny
Wielu naukowców uważa, że główną przyczyną tej choroby jest mutacja genetyczna, która prowadzi do gromadzenia się dużych ilości białka w komórkach. Wróżki i magowie twierdzą, że istnieją specjalne techniki wysyłania „obrażeń”, aby człowiek się zestarzał.
Nawiasem mówiąc, choroba ta występuje nie tylko u ludzi, ale także u zwierząt. Mają także cykle życia i okresy, które czasami przebiegają według scenariusza roku na trzy, a nawet dziesięć lat. Być może rozwiązanie problemu uda się znaleźć po wielu latach eksperymentów na naszych mniejszych braciach.
Jak ustalili naukowcy z Uniwersytetu Kalifornijskiego, lek zwany inhibitorem farnezylotransferazy znacząco zmniejsza częstość występowania objawów przedwczesnego starzenia się u myszy laboratoryjnych. Być może ten lek będzie odpowiedni do leczenia ludzi.
Oto jak kandydat nauk biologicznych Igor Bykow charakteryzuje objawy choroby u dzieci: „Progeria pojawia się nagle wraz z pojawieniem się dużych plam pigmentowych na ciele. Wtedy ludzie zaczynają cierpieć na prawdziwe choroby starcze. Rozwijają się u nich choroby serca, choroby naczyniowe, cukrzyca, wypadają włosy i zęby, a podskórna tkanka tłuszczowa zanika. Kości stają się łamliwe, skóra pomarszczona, a ciało garbione. Proces starzenia się u takich pacjentów zachodzi około dziesięciokrotnie szybciej niż u osoby zdrowej. Zło najprawdopodobniej jest zakorzenione w genach. Istnieje hipoteza, że nagle przestają wydawać komórkom polecenie podziału. I szybko stają się bezużyteczne.”
Geny przestają wydawać komórkom polecenie podziału, najwyraźniej ze względu na fakt, że końce DNA w chromosomach ulegają skróceniu – tak zwane telomery, których długość prawdopodobnie mierzy długość życia człowieka. Podobne procesy zachodzą u normalnych ludzi, ale znacznie wolniej. Nie jest jednak całkowicie jasne, w wyniku jakiego zaburzenia telomery ulegają skróceniu, a starzenie się zaczyna przyspieszać co najmniej 10-krotnie. Teraz naukowcy używają enzymów do wydłużania telomerów. Pojawiły się nawet doniesienia, że amerykańskim genetykom udało się w ten sposób przedłużyć życie much. Jednak wciąż jesteśmy daleko od wyników, które można zastosować w praktyce. Ludziom nie można pomóc nawet na poziomie eksperymentów. Na szczęście choroba nie jest dziedziczona.
Zakłada się, że nieprawidłowe działanie genomu występuje w okresie rozwoju wewnątrzmacicznego. Na razie nauka nie jest w stanie monitorować i zarządzać tą porażką: może jedynie stwierdzić fakt, ale być może w niedalekiej przyszłości gerontologia odpowie światu na to pytanie.
