
Alle Menschen werden alt. Laut Wissenschaftlern würden wir ohne die zerstörerischen Auswirkungen der äußeren Umgebung und unsere Sucht nach körperlich schädlichen Vergnügungen bis zu 130 oder sogar bis zu 150 Jahre alt werden. Und vor 16 Jahren, am 29. August 2001, gaben Wissenschaftler sogar bekannt, dass sie ein Langlebigkeitsgen gefunden hatten. Vielleicht können wir also in naher Zukunft die gesamte uns von der Natur vorgegebene Lebensspanne ausleben. Aber im Moment altern und sterben wir, die überwiegende Mehrheit vor dem 80. bis 90. Lebensjahr. Und manche Krankheiten verkürzen diesen ohnehin nicht so langen Zeitraum um ein Vielfaches. Und der „mörderischste“ unter ihnen ist im wahrsten Sinne des Wortes Progerie. MedAboutMe hat herausgefunden, wie es ist, in eineinhalb bis zwei Jahrzehnten alt zu werden.
Was ist Altern?

Altern ist ein natürlicher Prozess, der für jeden lebenden Organismus auf der Erde charakteristisch ist. Alle bestehenden Theorien zum Thema „Warum altern Menschen?“ lassen sich in zwei große Gruppen einteilen. Befürworter einer davon argumentieren, dass das Altern von der Natur für die weitere Entwicklung von Arten und Gesellschaft vorgesehen sei. Andere sind sich sicher, dass es hier keine globalen Pläne gibt – lediglich Schäden auf Gen- und Zellebene häufen sich mit der Zeit, was zu einer Abnutzung des Körpers führt.
Auf die eine oder andere Weise sammeln sich im Laufe des Lebens eines Menschen die Folgen innerer Fehler und Irrtümer sowie die Folgen äußerer Einflüsse in seinen Zellen und Geweben an. Zu den Schlüsselfaktoren des Alterns gehören:
- Exposition gegenüber reaktiven Sauerstoffspezies (ROS), die unser Körper natürlich braucht, aber nicht immer und nicht überall.
- Mutationen in der DNA somatischer Zellen (also Körperzellen). Das Genom ist keine in Zeit und Raum eingefrorene Struktur. Es ist eine lebendige und veränderliche Struktur.
- Ansammlung beschädigter Proteine, die ein Nebenprodukt von ROS oder Störungen von Stoffwechselprozessen sind.
- Verkürzung der Telomere – der Enden der Chromosomen. Zwar haben Wissenschaftler in letzter Zeit begonnen, daran zu zweifeln, dass Alterung mit Telomeren zusammenhängt, aber bisher ist diese Theorie immer noch populär.
Progerie, die in diesem Artikel besprochen wird, ist kein Altern – in dem Sinne, wie die Wissenschaft es versteht, wenn es um Lebenserwartung, Abnutzung des Körpers usw. geht. Diese Krankheit sieht aus wie Alterung, obwohl sie tatsächlich schwerwiegend ist eine genetische Erkrankung, die mit einer beeinträchtigten Produktion bestimmter Proteine einhergeht.
Progerie – Erkrankungen von Kindern und Erwachsenen
Der Engländer Jonathan Hutchinson beschrieb 1886 erstmals ein 6-jähriges Kind, bei dem er Hautatrophie beobachtete. Der Name der ungewöhnlichen Krankheit (vom griechischen Wort „progeros“ – alt vor ihrer Zeit) wurde ihr 1897 von Dr. Guilford gegeben, der die Nuancen der Krankheit untersuchte und beschrieb. Im Jahr 1904 veröffentlichte Dr. Werner eine Beschreibung der erwachsenen Progerie am Beispiel von vier Geschwistern, die an Katarakten und Sklerodermie litten.
Es wird vermutet, dass Francis Scott Fitzgerald seine Geschichte „Der seltsame Fall des Benjamin Button“ im Jahr 1922 genau unter dem Eindruck von Informationen über Progeriepatienten schrieb. Im Jahr 2008 spielte Brad Pitt den Protagonisten des Buches im Film „Der seltsame Fall des Benjamin Button“.
Es gibt zwei Arten von Progerie:
- Eine Krankheit, die Kinder betrifft, ist das Hutchinson-Gilford-Syndrom.
Dies ist eine seltene Pathologie. Es kommt bei einem von mehreren Millionen Kindern vor. Es wird angenommen, dass es heute auf der Welt nicht mehr als hundert Menschen gibt, die an Progerie im Kindesalter leiden. Wissenschaftler gehen jedoch davon aus, dass wir von etwa 150 weiteren nicht diagnostizierten Fällen sprechen können.
- Eine Krankheit, die Erwachsene betrifft, ist das Werner-Syndrom.
Dies ist ebenfalls eine seltene Krankheit, aber nicht so selten wie Progerie im Kindesalter. Menschen mit Werner-Syndrom werden in einem von 100.000 Fällen geboren. In Japan etwas häufiger: 1 Fall bei 20-40.000 Neugeborenen. Insgesamt sind weltweit etwas weniger als 1,5 Tausend solcher Patienten bekannt.
Gene und „Alter“

Progerie im Kindesalter hängt nur indirekt mit dem tatsächlichen Altern zusammen. Hierbei handelt es sich um eine Erkrankung aus der Gruppe der Laminopathien – Erkrankungen, die sich vor dem Hintergrund einer Störung der Produktion des Lamin-A-Proteins entwickeln. Wenn es nicht ausreicht oder der Körper das „falsche“ Lamin-A produziert, dann ein Ganzes Es entsteht eine Liste von Krankheiten, zu denen auch das Hutchinson-Gilford-Syndrom gehört.
Die Ursache für Progerie im Kindesalter ist eine Mutation im LMNA-Gen, das sich auf Chromosom 1 befindet. Dieses Gen kodiert für die Verbindung Prälamin A, aus der das Protein Lamin A gewonnen wird, das eine dünne Platte – die Lamina – bildet, die die innere Membran des Zellkerns bedeckt. Es ist für die Verankerung aller Arten von Molekülen und inneren Strukturen des Kerns notwendig. Fehlt Lamin A, kann das innere Gerüst des Zellkerns nicht aufgebaut werden, es kann seine Stabilität nicht aufrechterhalten, was zu einer beschleunigten Zerstörung der Zellen und des gesamten Organismus führt. Darüber hinaus spielt Lamin A eine Schlüsselrolle bei der Zellteilung. Es reguliert den Abbau und die Wiederherstellung des Zellkerns. Es ist nicht schwer, sich vorzustellen, was passieren kann, wenn dieses Protein fehlt oder nicht das ist, was es sein sollte. Eine Mutation des LMNA-Gens führt zur Bildung des „falschen“ Proteins – Progerin. Dies führt zu einer beschleunigten „Alterung“ von Kindern.
Den neuesten Daten zufolge tritt die Mutation in den frühen Stadien der Embryonalentwicklung auf und wird fast nie vom Elternteil auf das Kind übertragen.
Vor einigen Jahren entdeckten Wissenschaftler, dass Progerin auch von gesunden Zellen produziert wird, allerdings in deutlich geringeren Mengen als beim Hutchinson-Gilford-Syndrom. Darüber hinaus stellte sich heraus, dass mit zunehmendem Alter die Menge an Progerin in normalen Zellen zunimmt. Und das ist das Einzige, was die kindliche Progerie und den Alterungsprozess wirklich miteinander verbindet.
Progerie bei Erwachsenen ist das Ergebnis einer weiteren Mutation im WRN-Gen. Dieses Gen kodiert für ein Protein, das für die Aufrechterhaltung eines stabilen Zustands der Chromosomen notwendig ist, und ist auch an den Prozessen der Zellteilung beteiligt. Liegt eine Mutation im WRN-Gen vor, verändert sich die Struktur der Chromosomen ständig. Die Häufigkeit spontaner Mutationen steigt um das Zehnfache, während die Teilungsfähigkeit von Zellen im Vergleich zu gesunden Zellen um das Drei- bis Fünffache abnimmt. Auch die Telomerlänge nimmt ab. Und diese Prozesse kommen dem Altern, das wir meinen, wenn wir ältere Menschen auf einer Bank betrachten, schon sehr nahe.
Progerie bei Kindern

Bei Kindern manifestiert sich die Krankheit erstmals nach ein bis zwei Jahren. Diese Babys haben eine für ihr Alter unangemessene Kleinwüchsigkeit, einen großen Kopf mit einem charakteristisch reduzierten Gesichtsteil, eine dünne Nase, die wie ein Schnabel aussieht, und abstehende Ohren. Zu den charakteristischen Anzeichen gehören Mikrognathie (unterentwickelter Unterkiefer) und Exophthalmus (hervortretende Augen). Progerika haben eine sehr dünne Schicht aus Unterhautfettgewebe und dünne hyperpigmentierte Haut – sie trocknet schnell und bildet Falten, sieht durchscheinend, schlaff aus und hat durchscheinende Blutgefäße. Ihre Nägel sind entweder nicht vollständig entwickelt oder fehlen ganz. Haare werden schnell grau und fallen aus, selbst Wimpern und Augenbrauen sind spärlich und dünn. All dies verleiht ihnen das Aussehen kleiner alter Männer mit einem charakteristischen Aussehen. Daher der Mythos über schnell alternde Menschen.
Patienten mit Progerie entwickeln in der Regel eine verzögerte sexuelle Entwicklung und Tests zeigen erhöhte Cholesterin- und Lipoproteinwerte im Blut. Aber die Intelligenz der Progeriker leidet nicht. Die durchschnittliche Lebenserwartung dieser Kinder beträgt 13 Jahre. Ein Mann mit Progerie in der Kindheit, der 27 Jahre lebte, wurde zu einer „Langleberin“.
Manche Menschen mit Progerie in der Kindheit haben in ihrem kurzen Leben mehr erreicht als viele von uns. Leon Botha ist einer der langlebigen Progeriker; in den ihm zugeteilten 26 Jahren gelang es ihm, ein berühmter Künstler, Musiker und beliebter DJ zu werden. Und Sam Burns aus den USA, der 2014 im Alter von 17 Jahren starb, wurde als Motivationsredner und Aktivist im Bereich der Verbreitung von Informationen über seine Krankheit berühmt.
Die Haupttodesursachen bei Menschen mit Hutchinson-Gilford-Syndrom sind Arteriosklerose, die den gesamten Körper betrifft, und Myokardfibrose, der Ersatz des Herzmuskels durch Bindegewebe. Dabei lagert sich eine fettähnliche Substanz im Gehirn, in den Nieren, in der Leber, in den Eierstöcken (bei Frauen) und in den Hoden (bei Männern) ab und die Knochen werden dünner.
Progerie bei Erwachsenen
Bei Erwachsenen tritt das Werner-Syndrom erstmals im Alter zwischen 14 und 18 Jahren auf. Außerdem kommt es zu einer starken Gewichtsabnahme, Wachstumsstörungen, grauem Haar und Haarausfall. Ihr Körper verschlechtert sich rapide: Es werden Funktionsstörungen der Nieren, der Leber und der endokrinen Drüsen beobachtet, es entwickeln sich Osteoporose, Arteriosklerose, Diabetes mellitus, Katarakte und Herzinfarkt. Bei Männern und Frauen kommt es zu Veränderungen im Fortpflanzungssystem, die zu Unfruchtbarkeit führen. Bei jedem zehnten Menschen mit erwachsener Progerie wird im Alter von 30 bis 40 Jahren ein bösartiger Tumor diagnostiziert – Brustkrebs, Melanom, Sarkom usw. Solche Patienten werden etwa 45 bis 50 Jahre alt und sterben an Krebs oder Herzerkrankungen.
Im Gegensatz zur kindlichen Progerie leidet beim Werner-Syndrom auch die Psyche des Patienten.
Auf der Suche nach einem Heilmittel gegen vorzeitiges Altern

Es gibt noch keine Heilung für Progerie. Ärzte versuchen ihr Bestes, um das schnelle „Altern“ dieser Patienten zu verlangsamen. Kindern mit Hutchinson-Gilford-Syndrom werden Medikamente mit antioxidativen Eigenschaften, Vitamin E usw. verschrieben. Sie unterziehen sich einer Koronararterien-Bypass-Operation, um Komplikationen im Herzen und in den Blutgefäßen zu bewältigen.
Ein Medikament, das das Leben eines Patienten mit Progerie im Kindesalter um ein Jahr, bestenfalls um 6-7 Jahre verlängern kann, ist Lonafarnib, ein Medikament zur Behandlung von Krebs. Für eine Person, deren Leben im Alter von 13 bis 17 Jahren enden kann, ist dies ein bedeutender Zeitraum. Im Jahr 2012 wurde festgestellt, dass die Einnahme zu einem verbesserten Hörvermögen, stärkeren Knochen, einem erhöhten Körpergewicht und einer erhöhten Gefäßflexibilität führt. 6 Jahre sind nicht so lang, aber die Entwicklung vieler wirksamer Medikamente begann mit ähnlichen Erfolgen.
Ermutigende Ergebnisse wurden auch bei der Therapie mit folgenden Medikamenten erzielt:
Rapamycin, ein Immunsuppressivum, das die Lebensdauer der Muskeln von Kindern mit Progerie verlängert;
Pravastatin, das den Cholesterinspiegel senkt;
Zoledronat, das die Entwicklung von Knochenmetastasen bei Krebs verlangsamt.
Ist Progerieprävention notwendig? Früher wurde angenommen, dass dies nicht der Fall sei, da Menschen mit Progerie im Kindesalter nicht lange genug lebten, um Kinder zur Welt zu bringen, und erwachsene Patienten Unfruchtbarkeit entwickelten. Zwar gab es in den letzten Jahren Berichte über Frauen mit Progerie, die gesunde Kinder zur Welt brachten. Diese Krankheit ist jedoch so selten, dass die Wahrscheinlichkeit einer Mutationsausbreitung äußerst gering ist.
Für Progerie bei Erwachsenen wurde noch keine Heilung gefunden. Bei solchen Patienten werden nur die schweren und zahlreichen Krankheiten behandelt, die sie befallen.
Die Wissenschaft steht nicht still, insbesondere im Bereich der Medizin. Unsere Vorfahren konnten sich nicht einmal vorstellen, dass Krankheiten wie Schwindsucht (Tuberkulose) oder Pest in Zukunft heilbar sein würden. Aber trotz dieses Fortschritts gibt es in der modernen Welt 15.000 Krankheiten. Und während ein Mensch mit vielen Virus- oder Infektionskrankheiten zurechtkommt, sind einige genetische Mutationen nicht heilbar. Wir stellen Ihnen die 10 ungewöhnlichsten Krankheiten der Welt vor.
1. Progerie oder vorzeitiges Alterungssyndrom
- Anzahl der Personen, bei denen die Krankheit diagnostiziert wurde:

Kind mit Progerie-Krankheit
Ein Jahr im Leben einer solchen Person kann als 10-15 Jahre im Leben eines gewöhnlichen Menschen angesehen werden, sodass Kinder in 90 % der Fälle vor Erreichen des 15. Lebensjahres an dieser Krankheit sterben. Todesursachen sind meist Herzinfarkte oder multiple Blutungen.
2. Kleine-Levin-Syndrom
- Anzahl der Menschen, bei denen die Krankheit diagnostiziert wurde: 20 weltweit
- Durchschnittliche Lebenserwartung: gleich wie bei normalen Menschen
- Gründe: unbekannt
- Behandlung: unheilbar
Beim Kleine-Levin-Syndrom (auch bekannt als „Dornröschenkrankheit“) kommt es bei Patienten regelmäßig zu übermäßigen Schlafanfällen (ab 18 Stunden pro Tag), die zwischen 2 und 40 Tagen dauern und nur zur Befriedigung biologischer Bedürfnisse unterbrochen werden.
Es ist äußerst schwierig, einen solchen Menschen während des „Winterschlafes“ aufzuwecken, und nach dem Aufwachen wird er äußerst aggressiv. Bei der Rückkehr zum normalen Lebensrhythmus erfährt der Patient eine Amnesie für die Ereignisse während des Anfalls. In 90 % der Fälle tritt sie bei Jugendlichen auf und ist unheilbar, kann jedoch mit zunehmendem Alter von selbst verschwinden.
3. Fibrodysplasie oder Stone-Man-Syndrom
- Anzahl der Menschen, bei denen die Krankheit diagnostiziert wurde: 500 weltweit
- Durchschnittliche Lebenserwartung: 30 Jahre
- Ursache: Mutationen auf Genebene
- Behandlung: unheilbar
Auch Prellungen sind für Menschen mit Fibrodysplasie gefährlich: An der Verletzungsstelle (Schlag, Hämatom oder Prellung) kommt es beim Patienten zur Bildung von neuem Knochengewebe, einem „zweiten Skelett“.
Häufige Schäden am Muskelgewebe können zur vollständigen Immobilisierung einer Person führen. Die moderne Medizin hat keine Methoden zur Behandlung dieser Krankheit erfunden, aber amerikanische Wissenschaftler haben eine Reihe erfolgreicher Studien durchgeführt, die den Patienten Hoffnung auf eine gesunde Zukunft geben.
4. Porphyrie
- Ursache: Störung der Synthese roter Blutkörperchen, Erbkrankheit
- Behandlung: Einnahme von Hormonpräparaten, Hepatoprotektoren, Transfusion roter Blutkörperchen.
Porphyrie oder Vampirismus ist eine genetische Erkrankung, bei der die Haut einer Person bei Kontakt mit Sonnenlicht zu jucken beginnt, Narben bildet, Blasen bildet und aufplatzt.

Frau mit Vampirismus-Syndrom (Porphyrie)
Die Krankheit betrifft jedoch nicht nur die Haut: Nägel, Zahnfleisch und Ohren der Patienten sind deformiert; Zähne und Zahnfleisch werden bei Sonneneinstrahlung rot und die Hornhaut der Augen wird trüb. Menschen mit Porphyrie verlassen das Haus am liebsten abends oder nachts, wenn keine Sonne scheint. Vielleicht sind aus diesem Grund Legenden über Vampire entstanden.
5. Mobius-Syndrom
- Ursachen: unbekannt, die Erkrankung beruht auf einer Lähmung oder Unterentwicklung des Gesichtsnervs.
- Behandlung: Es gibt keine radikalen Maßnahmen zur Heilung dieses Syndroms; Patienten werden bereits in der frühen Kindheit einer Korrektur von Schielen, Sprache usw. unterzogen. Die ausgeprägtesten Defekte werden durch eine Operation beseitigt.

Mädchen mit Möbius-Syndrom
Diese angeborene Anomalie ist durch eine abnormale Entwicklung oder Lähmung des Gesichtsnervs gekennzeichnet, wodurch die Mimik im Gesicht der Person ganz oder teilweise fehlt. Menschen mit diesem Syndrom können nicht lachen und haben sogar Schwierigkeiten beim Schlucken; ihr Gesicht sieht eher aus wie eine Maske.
6. Epidermolysis bullosa oder Schmetterlingskrankheit
- Anzahl der Personen, bei denen die Krankheit diagnostiziert wurde: unbekannt
- Durchschnittliche Lebenserwartung: 30 Jahre
- Ursache: Chromosomenmutation
- Behandlung: unheilbar
Schwimmen, Sportspiele, Umarmungen – das ist eine kurze Liste von Dingen, die für „Schmetterlingsmenschen“ unzugänglich sind.

Epidermolysis bullosa oder Schmetterlingskrankheit
Epidermolysis bullosa zeichnet sich durch eine hohe Empfindlichkeit der Haut aus, wodurch sich bei Patienten mit der geringsten mechanischen Schädigung (sogar Reibung) der Haut an der Verletzungsstelle Blasen und Geschwüre bilden, die der Person starke Schmerzen bereiten.
Die Krankheit selbst ist nicht tödlich, aber 90 % der Kinder unter 3 Jahren sterben an unsachgemäßer Pflege, der Rest muss lebenslang schreckliche Schmerzen ertragen, da die Krankheit unheilbar ist.
7. Hypertrichose oder Wolfssyndrom
- Ursachen: Genmutationen/Funktionsstörungen der endokrinen Drüsen oder hormonelles Ungleichgewicht
- Behandlung: Die angeborene Form der Erkrankung ist unheilbar; bei der erworbenen Form erfolgt die Behandlung durch einen Endokrinologen,

Hypertrichose oder Wolfssyndrom
Diese Krankheit ist typisch für Kinder im frühen Alter und zeichnet sich durch einen übermäßigen Haarwuchs an verschiedenen Körperstellen aus, der nicht dem Alter/Geschlecht der Person entspricht. Dieser abnormale Haarwuchs lässt Menschen wie Tiere aussehen. Angeborene Hypertrichose ist zwar nicht gesundheitsschädlich, aber auch nicht behandelbar. Patienten müssen ständig auf Haarentfernungsdienste zurückgreifen, da diese Verstöße unästhetisch wirken.
8. Elephantiasis oder Elephantiasis
- Ursachen: Parasiten, die Filariose übertragen/abnormale Entwicklung des Lymphsystems
- Behandlung: Antibiotika mit Avermectin.
Bei dieser Krankheit bildet sich aufgrund der Stagnation der Lymphe in den Gliedmaßen und der Störung ihrer Eigenschaften an der Ödemstelle neues Bindegewebe, wodurch die Gliedmaße um ein Vielfaches größer wird.
Die Gliedmaßen mit Elephantiasis ähneln denen von Elefanten und die betroffenen Hautbereiche sind mit Rissen, Geschwüren und Warzen bedeckt. Solche Veränderungen hindern eine Person daran, sich normal zu bewegen, anstrengenden Aktivitäten nachzugehen und sich sogar zu waschen. Die Krankheit ist mit neuen Antibiotika behandelbar, die 2015 entdeckt wurden.
9. Aquagene Urtikaria oder Allergie gegen Wasser
- Anzahl der Menschen, bei denen die Krankheit diagnostiziert wurde: 50 weltweit
- Gründe: unbekannt
- Behandlung: unheilbar; Die Symptome werden durch Antihistaminika gelindert
Allergie gegen Wasser oder aquagene Urtikaria
Bei Kontakt mit Wasser verspürt die Haut einer erkrankten Person Schmerzen und an den betroffenen Stellen treten Hautausschläge auf, begleitet von Juckreiz und Peeling. Diese Krankheit ist nicht tödlich, schränkt aber das Leben erkrankter Menschen stark ein: Auf Regen, Tränen und sogar Schweiß kommt es zu einer allergischen Reaktion.
10. Cotard-Syndrom oder Living-Corpse-Syndrom
- Ursachen: Depression, psychische Störungen
- Behandlung: Einnahme von Antidepressiva und Psychopharmaka, Beobachtung durch einen Psychiater
Menschen, die an diesem Syndrom leiden, sind davon überzeugt, dass sie tot und nur lebende Leichen sind. Sie riechen die Verrottung ihrer inneren Organe und wie Würmer sie von innen fressen. Die Patienten sind zuversichtlich, dass sie weder Nahrung noch Wasser benötigen. Diese Störung geht mit schweren Depressionen und Selbstmordtendenzen einher.
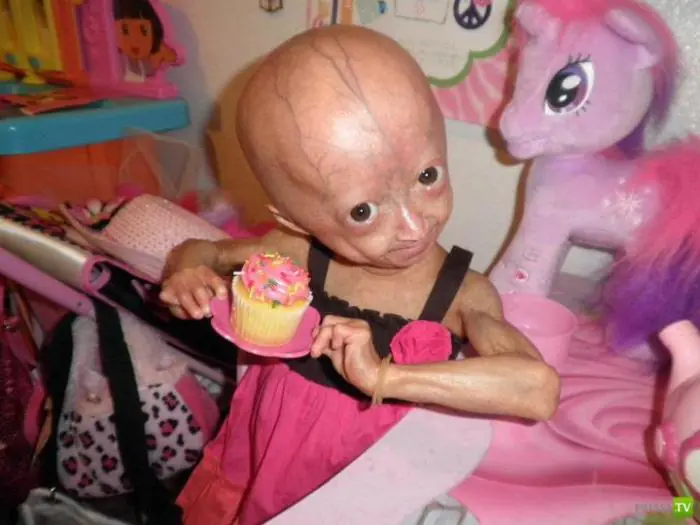
Im Oktober 2005 führten Ärzte in einer Moskauer Klinik die erste Operation an einem Patienten durch, der an einem vorzeitigen Alterungssyndrom litt. Progerie ist eine sehr seltene Krankheit. Medizinische Koryphäen auf der ganzen Welt behaupten, dass Menschen von dem Moment an, in dem diese Krankheit im Körper „erwacht“, im Durchschnitt nur 13 Jahre leben.
Laut Statistik wird etwa einer von vier Millionen Menschen mit einem solchen Gendefekt geboren. Progerie wird in Progerie im Kindesalter, Hutchinson-Gilford-Syndrom genannt, und Progerie im Erwachsenenalter, Werner-Syndrom genannt, unterteilt. In beiden Fällen bricht der genetische Mechanismus zusammen und es beginnt eine unnatürliche Erschöpfung aller lebenserhaltenden Systeme. Beim Hutchinson-Gilford-Syndrom ist die körperliche Entwicklung der Kinder verzögert, während gleichzeitig in den ersten Lebensmonaten Anzeichen von Altersergrauung, Haarausfall und Falten auftreten.
Mit fünf Jahren leidet ein solches Kind an allen Altersbeschwerden: Hörverlust, Arthritis, Arteriosklerose und wird nicht einmal 13 Jahre alt. Beim Werner-Syndrom beginnen junge Menschen im Alter von 16 bis 20 Jahren schnell zu altern, und im Alter von 30 bis 40 Jahren sterben solche Patienten mit allen Symptomen extremen Alters.
Es gibt keine Heilung für Progerie – mit allen wissenschaftlichen Fortschritten kann man den irreversiblen Prozess nur verlangsamen.
Gestohlene Jugend
Fälle von plötzlichem Altern sind sehr prosaisch: Ein Kind, das unter normalen Bedingungen lebt, überrascht seine Umgebung zunächst mit seiner schnellen Entwicklung. In jungen Jahren sieht er aus wie ein Erwachsener, und dann zeigt er alle Anzeichen des … nahenden Alters.

Im Jahr 1716 starb in der englischen Stadt Nottingham der achtzehnjährige Sohn von Earl William of Sheffield, der im Alter von dreizehn Jahren zu altern begann. Der junge Sheffield sah viel älter aus als sein Vater: graue Haare, halb verlorene Zähne, faltige Haut. Der unglückliche junge Mann sah aus wie ein vom Leben gebeutelter Mann, er litt sehr darunter und akzeptierte den Tod als Erlösung von der Qual.
Fälle dieser Art gibt es bei Vertretern königlicher Familien. Der ungarische König Ludwig II. war im Alter von neun Jahren bereits in der Pubertät und vergnügte sich gerne mit den Hofmädchen. Mit vierzehn bekam er einen dicken Vollbart und sah aus, als wäre er mindestens 35 Jahre alt. Ein Jahr später heiratete er und an seinem sechzehnten Geburtstag schenkte ihm seine Frau einen Sohn. Doch im Alter von achtzehn Jahren wurde Ludwig völlig ergraut und starb zwei Jahre später mit allen Anzeichen alter Altersschwäche.
Es ist merkwürdig, dass weder der Königssohn noch seine weiteren Nachkommen eine solche Krankheit geerbt haben. Unter den Beispielen des 19. Jahrhunderts kann man die Geschichte eines einfachen Dorfmädchens, der Französin Louise Ravaillac, hervorheben. Im Alter von acht Jahren wurde Louise, als Frau voll ausgebildet, von einem örtlichen Hirten schwanger und brachte ein völlig gesundes Kind zur Welt. Mit ihrem sechzehnten Geburtstag hatte sie bereits drei Kinder und sah älter aus als ihre Mutter; mit 25 Jahren verwandelte sie sich in eine altersschwache alte Frau und starb noch vor ihrem 26. Lebensjahr an Altersschwäche.
Nicht weniger interessant sind die Schicksale der Menschen, die im 20. Jahrhundert lebten. Einige von ihnen hatten etwas mehr Glück als andere. Michael Sommers zum Beispiel, ein Einwohner der amerikanischen Stadt San Bernardino, geboren 1905, wurde früh erwachsen und alterte und konnte 31 Jahre alt werden. Der superschnelle Einstieg ins Erwachsenenalter gefiel ihm zunächst sogar. Doch als Michael mit siebzehn Jahren mit Entsetzen feststellte, dass er zu altern begann, begann er verzweifelte Versuche, diesen zerstörerischen Prozess zu stoppen.
Aber die Ärzte zuckten nur mit den Schultern und konnten nichts tun, um zu helfen. Nachdem Sommers dauerhaft ins Dorf gezogen war und begonnen hatte, viel Zeit an der frischen Luft zu verbringen, gelang es Sommers, seine Schwäche ein wenig zu verlangsamen. Dennoch verwandelte er sich im Alter von 30 Jahren in einen alten Mann und ein Jahr später wurde er von einer gewöhnlichen Grippe getötet. Unter anderen ähnlichen Phänomenen kann man die Engländerin Barbara Dahlin hervorheben, die 1982 im Alter von 26 Jahren starb.
Als Barbara im Alter von 20 Jahren verheiratet war und zwei Kinder zur Welt brachte, alterte sie schnell und unwiderruflich. Deshalb verließ sie ihr junger Mann, der nicht mit dem „alten Wrack“ leben wollte. Im Alter von 22 Jahren wurde die „alte Frau“ aufgrund ihres sich verschlechternden Gesundheitszustands und der erlittenen Schocks blind und bewegte sich bis zu ihrem Tod durch Berührung oder in Begleitung eines Blindenhundes, den ihr die Behörden ihrer Heimatstadt Birmingham gegeben hatten.
Paul Demongeau aus der französischen Stadt Marseille ist 23 Jahre alt. Gleichzeitig sieht er aus wie 60 und fühlt sich wie ein alter Mann. Allerdings hat er die Hoffnung noch nicht verloren, dass ein Wunder geschieht und ein Heilmittel gefunden wird, das seinem raschen Verfall Einhalt gebietet. Sein unglücklicher Bruder, ein Sizilianer aus der Stadt Syrakus, Mario Termini, ist noch nicht einmal 20 Jahre alt, sieht aber viel älter als 30 aus. Termini, der Sohn wohlhabender Eltern, verweigert sich nichts, trifft sich mit lokalen Schönheiten und führt ein ausgelassener Lebensstil.
Was haben wir?
In unserem Land lebten auch „frühreife“ Menschen. Noch zur Zeit Iwans des Schrecklichen starb der Sohn der Michailow-Bojaren, Wassili, im Alter von 19 Jahren als altersschwacher Mann. 1968 starb Nikolai Schorikow, ein Arbeiter in einer der Fabriken, im Alter von 22 Jahren in Swerdlowsk. Mit sechzehn Jahren begann er zu altern, was die Ärzte sehr verwirrte. Die Koryphäen der Medizin zuckten nur mit den Schultern: „Das kann doch nicht sein!“
Als alter Mann in einem Alter, in dem alles gerade erst anfängt, verlor Nikolai jegliches Interesse am Leben und beging Selbstmord, indem er Tabletten schluckte... Und dreizehn Jahre später starb der 28-jährige „alte Mann“ Sergei Efimov in Leningrad. Seine Jugendzeit endete im Alter von elf Jahren, mit zwanzig Jahren begann er merklich zu altern und starb als altersschwacher alter Mann, nachdem er ein Jahr vor seinem Tod die Fähigkeit zu vernünftigem Denken fast vollständig verloren hatte.
Gene sind an allem schuld
Viele Wissenschaftler glauben, dass die Hauptursache dieser Krankheit eine genetische Mutation ist, die zur Ansammlung großer Proteinmengen in Zellen führt. Hellseher und Magier behaupten, dass es spezielle Techniken gibt, um „Schaden“ zu verursachen, um einen Menschen altern zu lassen.
Diese Krankheit kommt übrigens nicht nur beim Menschen, sondern auch bei Tieren vor. Sie haben auch Lebenszyklen und Zeiträume, die manchmal dem Drehbuch eines Jahres in drei oder sogar zehn Jahren folgen. Vielleicht wird nach jahrelangen Experimenten an unseren kleineren Brüdern eine Lösung für das Problem gefunden.
Wie Forscher der University of California herausgefunden haben, reduziert ein Medikament namens Farnesyltransferase-Inhibitor die Rate der Symptome vorzeitiger Alterung bei Labormäusen deutlich. Vielleicht eignet sich dieses Arzneimittel zur Behandlung von Menschen.
So charakterisiert der Kandidat der Biowissenschaften Igor Bykov die Krankheitssymptome bei Kindern: „Progerie tritt plötzlich mit dem Auftreten großer Pigmentflecken am Körper auf. Dann beginnen die Menschen an echten Alterskrankheiten zu leiden. Sie entwickeln Herzerkrankungen, Gefäßerkrankungen, Diabetes, Haare und Zähne fallen aus und das Unterhautfettgewebe verschwindet. Knochen werden brüchig, die Haut wird faltig und der Körper krümmt sich. Der Alterungsprozess verläuft bei solchen Patienten etwa zehnmal schneller als bei einem gesunden Menschen. Das Böse wurzelt höchstwahrscheinlich in den Genen. Es gibt eine Hypothese, dass sie plötzlich aufhören, den Zellen den Befehl zur Teilung zu geben. Und sie werden schnell unbrauchbar.“
Gene geben den Zellen keinen Befehl mehr, sich zu teilen, was offenbar daran liegt, dass die Enden der DNA in den Chromosomen verkürzt werden – die sogenannten Telomere, deren Länge vermutlich die Länge eines menschlichen Lebens misst. Bei normalen Menschen laufen ähnliche Prozesse ab, allerdings deutlich langsamer. Es ist jedoch völlig unklar, durch welche Störung sich die Telomere verkürzen und die Alterung um mindestens das Zehnfache beschleunigt. Jetzt nutzen Wissenschaftler Enzyme, um Telomere zu verlängern. Es gab sogar Berichte, dass es amerikanischen Genetikern auf diese Weise gelungen sei, das Leben von Fliegen zu verlängern. Doch von praxistauglichen Ergebnissen sind wir noch weit entfernt. Selbst auf der Ebene von Experimenten kann den Menschen nicht geholfen werden. Glücklicherweise wird die Krankheit nicht vererbt.
Es wird angenommen, dass es während der intrauterinen Entwicklung zu einer Fehlfunktion des Genoms kommt. Bisher kann die Wissenschaft dieses Versagen nicht überwachen und bewältigen: Sie kann nur eine Tatsache feststellen, aber vielleicht wird die Gerontologie in naher Zukunft der Welt diese Frage beantworten.
